

This is a preprint.
Long-read sequencing of hundreds of diverse brains provides insight into the impact of structural variation on gene expression and DNA methylation
- PMID: 39764002
- PMCID: PMC11702628
- DOI: 10.1101/2024.12.16.628723
Long-read sequencing of hundreds of diverse brains provides insight into the impact of structural variation on gene expression and DNA methylation
Abstract
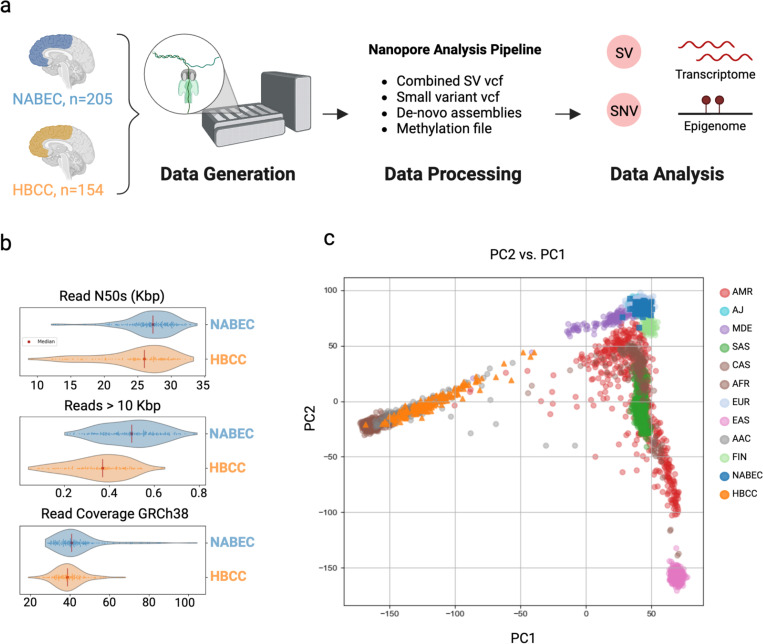
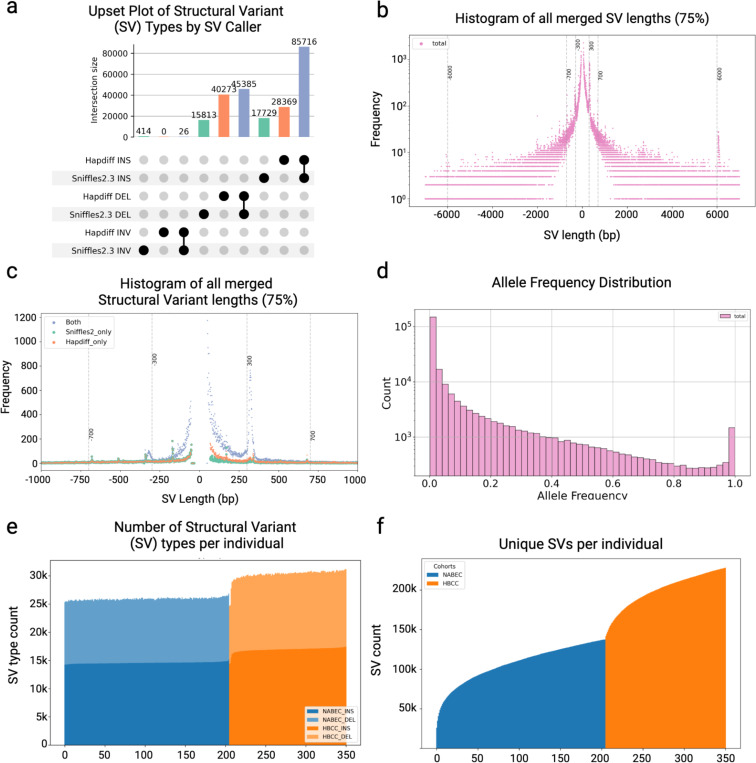
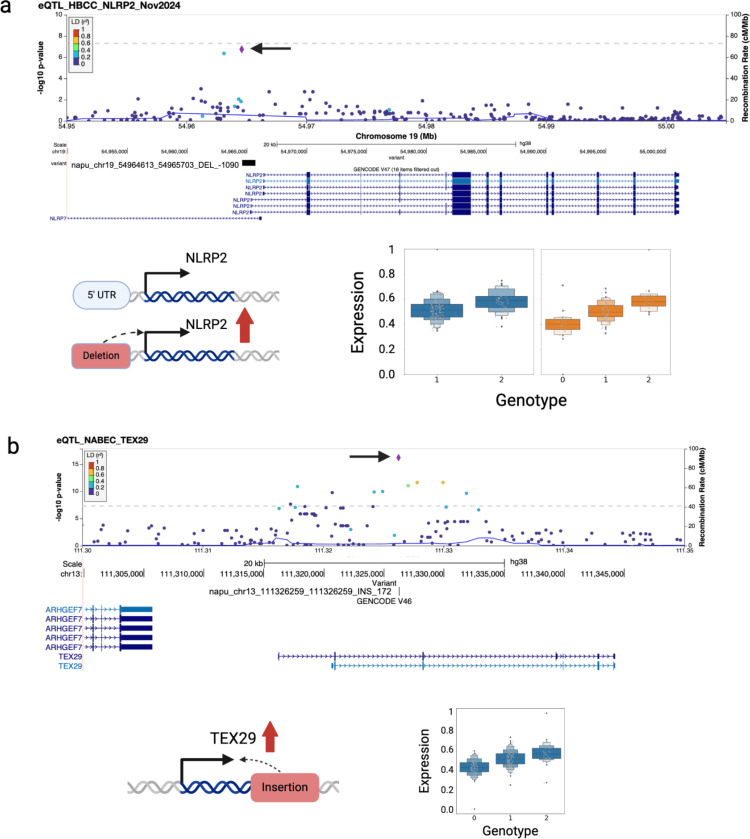
Structural variants (SVs) drive gene expression in the human brain and are causative of many neurological conditions. However, most existing genetic studies have been based on short-read sequencing methods, which capture fewer than half of the SVs present in any one individual. Long-read sequencing (LRS) enhances our ability to detect disease-associated and functionally relevant structural variants (SVs); however, its application in large-scale genomic studies has been limited by challenges in sample preparation and high costs. Here, we leverage a new scalable wet-lab protocol and computational pipeline for whole-genome Oxford Nanopore Technologies sequencing and apply it to neurologically normal control samples from the North American Brain Expression Consortium (NABEC) (European ancestry) and Human Brain Collection Core (HBCC) (African or African admixed ancestry) cohorts. Through this work, we present a publicly available long-read resource from 351 human brain samples (median N50: 27 Kbp and at an average depth of ~40x genome coverage). We discover approximately 234,905 SVs and produce locally phased assemblies that cover 95% of all protein-coding genes in GRCh38. Utilizing matched expression datasets for these samples, we apply quantitative trait locus (QTL) analyses and identify SVs that impact gene expression in post-mortem frontal cortex brain tissue. Further, we determine haplotype-specific methylation signatures at millions of CpGs and, with this data, identify cis-acting SVs. In summary, these results highlight that large-scale LRS can identify complex regulatory mechanisms in the brain that were inaccessible using previous approaches. We believe this new resource provides a critical step toward understanding the biological effects of genetic variation in the human brain.
Conflict of interest statement
K.S. is an employee of Google LLC and owns Alphabet stock as part of the standard compensation package; authors from Google LLC did not have access to the cell line and brain tissue sample data. WT has two patents (8,748,091 and 8,394,584) licensed to Oxford Nanopore Technologies. F.J.S. received research support from Illumina, Pacific Biosciences and Oxford Nanopore Technologies. DEM is on a scientific advisory board at Oxford Nanopore Technologies (ONT), is engaged in a research agreement with ONT, and they have paid for him to travel to speak on their behalf. DEM is a scientific advisory board member at Basis Genetics. DEM holds stock options in MyOme and Basis Genetics. This research was supported in part by the Intramural Research Program of the NIH, National Institute on Aging (NIA), National Institutes of Health, Department of Health and Human Services; project number ZO1 AG000534. This work utilized the computational resources of the NIH STRIDES Initiative (https://cloud.nih.gov) through the Other Transaction agreement - Azure: OT2OD032100, Google Cloud Platform: OT2OD027060, Amazon Web Services: OT2OD027852. This work utilized the computational resources of the NIH HPC Biowulf cluster (https://hpc.nih.gov). Some authors’ participation in this project was part of a competitive contract awarded to DataTecnica LLC by the National Institutes of Health to support open science research. M.A.N. also owns stock in Character Bio Inc. and Neuron23 Inc.
Figures



References
Publication types
Grants and funding
- U01 HG010961/HG/NHGRI NIH HHS/United States
- P30 AG072946/AG/NIA NIH HHS/United States
- T32 HG012344/HG/NHGRI NIH HHS/United States
- P30 AG072980/AG/NIA NIH HHS/United States
- OT2 OD033761/OD/NIH HHS/United States
- OT2 OD027060/OD/NIH HHS/United States
- R01 HG010485/HG/NHGRI NIH HHS/United States
- U24 NS072026/NS/NINDS NIH HHS/United States
- P30 AG019610/AG/NIA NIH HHS/United States
- ZIA AG000538/ImNIH/Intramural NIH HHS/United States
- OT2 OD032100/OD/NIH HHS/United States
- OT3 HL142481/HL/NHLBI NIH HHS/United States
- U24 HG010262/HG/NHGRI NIH HHS/United States
- OT2 OD027852/OD/NIH HHS/United States
- ZIC MH002903/ImNIH/Intramural NIH HHS/United States
- ZIA AG000534/ImNIH/Intramural NIH HHS/United States
- U24 HG011853/HG/NHGRI NIH HHS/United States
- DP5 OD033357/OD/NIH HHS/United States
LinkOut - more resources
Full Text Sources
Miscellaneous