

This is a preprint.
Isobaric Tagging and Data Independent Acquisition as Complementary Strategies for Proteome Profiling on an Orbitrap Astral Mass Spectrometer
- PMID: 39764012
- PMCID: PMC11702835
- DOI: 10.1101/2024.12.17.628765
Isobaric Tagging and Data Independent Acquisition as Complementary Strategies for Proteome Profiling on an Orbitrap Astral Mass Spectrometer
Update in
-
Isobaric Tagging and Data Independent Acquisition as Complementary Strategies for Proteome Profiling on an Orbitrap Astral Mass Spectrometer.J Proteome Res. 2025 Mar 7;24(3):1414-1424. doi: 10.1021/acs.jproteome.4c01107. Epub 2025 Feb 12. J Proteome Res. 2025. PMID: 39937051
Abstract
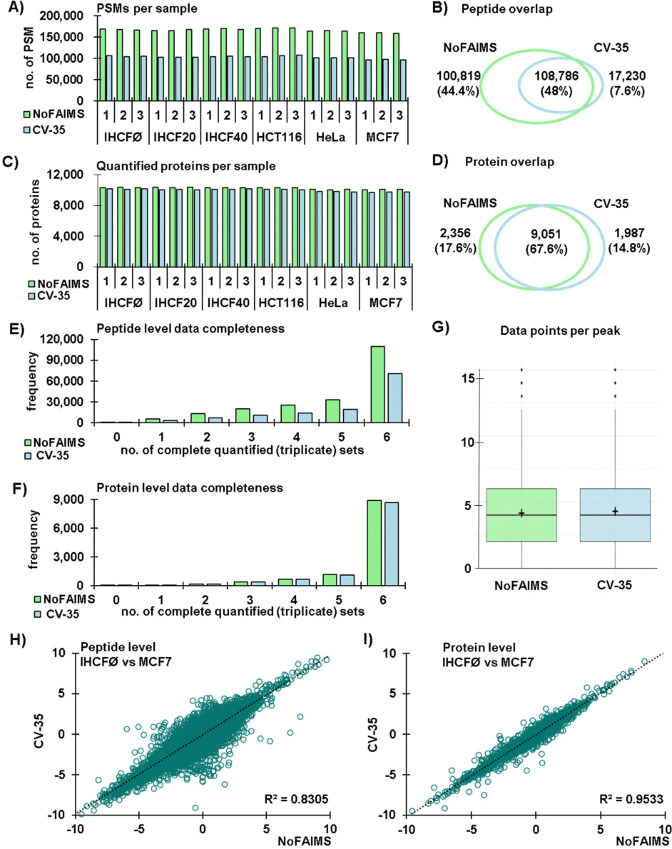
Comprehensive global proteome profiling that is amenable to high throughput processing will broaden our understanding of complex biological systems. Here, we evaluated two leading mass spectrometry techniques, Data Independent Acquisition (DIA) and Tandem Mass Tagging (TMT), for extensive protein abundance profiling. DIA provides label-free quantification with a broad dynamic range, while TMT enables multiplexed analysis using isobaric tags for efficient cross-sample comparisons. We analyzed 18 samples, including four cell lines (IHCF, HCT116, HeLa, MCF7) under standard growth conditions, in addition to IHCF treated with two H2O2 concentrations, all in triplicate. Experiments were conducted on an Orbitrap Astral mass spectrometer, employing Field Asymmetric Ion Mobility Spectrometry (FAIMS). Despite utilizing different acquisition strategies, both the DIA and TMT approaches achieved comparable proteome depth and quantitative consistency, with each method quantifying over 10,000 proteins across all samples, with slightly more protein-level precision for the TMT strategy. Relative abundance correlation analysis showed strong agreement at both peptide and protein levels. Our findings highlight the complementary strengths of DIA and TMT for high-coverage proteomic studies, providing flexibility in method selection based on specific experimental needs.
Keywords: Astral; DIA; FAIMS; IHCF; TMTpro.
Figures



References
-
- Gillet L.C., Navarro P., Tate S., Rost H., Selevsek N., Reiter L., Bonner R., Aebersold R., Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis, Mol Cell Proteomics 11(6) (2012) O111 016717. - PMC - PubMed
-
- Thompson A., Schafer J., Kuhn K., Kienle S., Schwarz J., Schmidt G., Neumann T., Johnstone R., Mohammed A.K., Hamon C., Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS, Anal Chem 75(8) (2003) 1895–904. - PubMed
-
- Ross P.L., Huang Y.N., Marchese J.N., Williamson B., Parker K., Hattan S., Khainovski N., Pillai S., Dey S., Daniels S., Purkayastha S., Juhasz P., Martin S., Bartlet-Jones M., He F., Jacobson A., Pappin D.J., Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents, Mol Cell Proteomics 3(12) (2004) 1154–69. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources