

This is a preprint.
Same-Slide Spatial Multi-Omics Integration Reveals Tumor Virus-Linked Spatial Reorganization of the Tumor Microenvironment
- PMID: 39764057
- PMCID: PMC11702642
- DOI: 10.1101/2024.12.20.629650
Same-Slide Spatial Multi-Omics Integration Reveals Tumor Virus-Linked Spatial Reorganization of the Tumor Microenvironment
Abstract
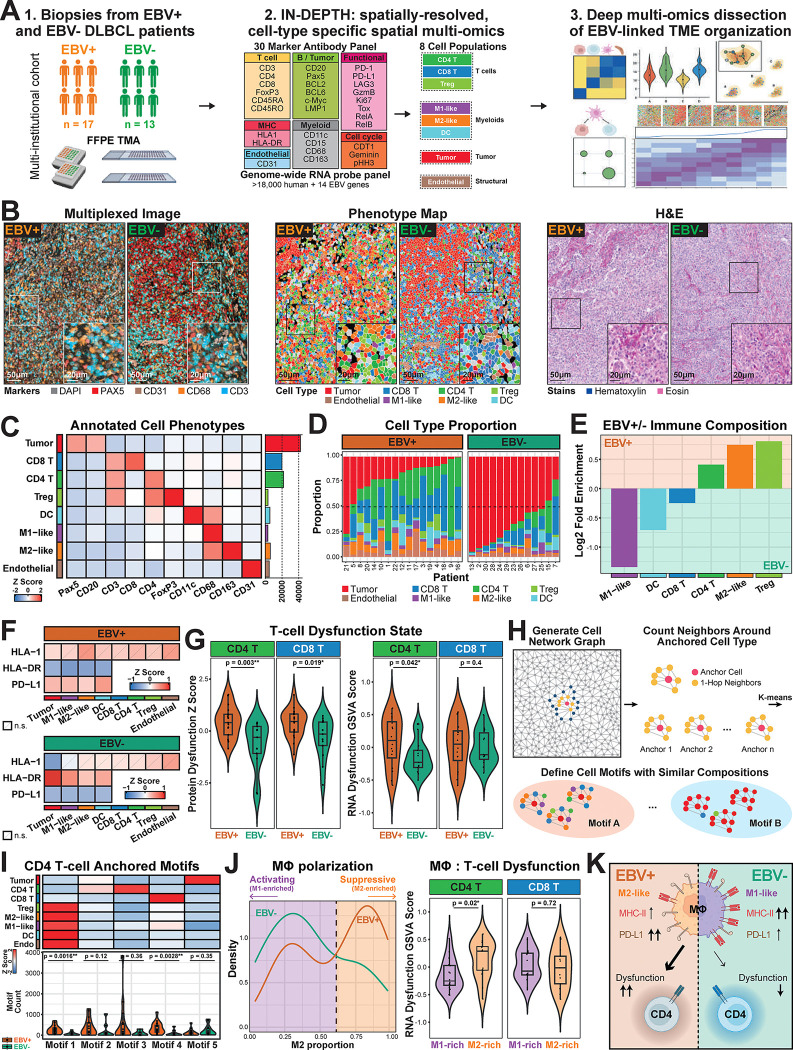
The advent of spatial transcriptomics and spatial proteomics have enabled profound insights into tissue organization to provide systems-level understanding of diseases. Both technologies currently remain largely independent, and emerging same slide spatial multi-omics approaches are generally limited in plex, spatial resolution, and analytical approaches. We introduce IN-situ DEtailed Phenotyping To High-resolution transcriptomics (IN-DEPTH), a streamlined and resource-effective approach compatible with various spatial platforms. This iterative approach first entails single-cell spatial proteomics and rapid analysis to guide subsequent spatial transcriptomics capture on the same slide without loss in RNA signal. To enable multi-modal insights not possible with current approaches, we introduce k-bandlimited Spectral Graph Cross-Correlation (SGCC) for integrative spatial multi-omics analysis. Application of IN-DEPTH and SGCC on lymphoid tissues demonstrated precise single-cell phenotyping and cell-type specific transcriptome capture, and accurately resolved the local and global transcriptome changes associated with the cellular organization of germinal centers. We then implemented IN-DEPTH and SGCC to dissect the tumor microenvironment (TME) of Epstein-Barr Virus (EBV)-positive and EBV-negative diffuse large B-cell lymphoma (DLBCL). Our results identified a key tumor-macrophage-CD4 T-cell immunomodulatory axis differently regulated between EBV-positive and EBV-negative DLBCL, and its central role in coordinating immune dysfunction and suppression. IN-DEPTH enables scalable, resource-efficient, and comprehensive spatial multi-omics dissection of tissues to advance clinically relevant discoveries.
Keywords: Bioinformatics; Computational Biology; DLBCL; EBV; Graph Signal Processing; Spatial Multi-Omics; Spatial Proteomics; Spatial Transcriptomics; Systems Immunology; Tumor Microenvironment; Tumor Virus.
Conflict of interest statement
CONFLICT OF INTERESTS S.J. is a co-founder of Elucidate Bio Inc, has received speaking honorariums from Cell Signaling Technology, and has received research support from Roche and Sanofi unrelated to this w ork. S.J.R. has received research support from Affimed, Merck, and Bristol-Myers Squibb (BMS), is on the Scientific Advisory Board for Immunitas Therapeutics, and also a part of the BMS International Immuno-Oncology Network (II-ON) unrelated to this work. F.S.H. has leadership roles at Bicara Therapeutics, stock and ownership interests in Apricity Health, Torque, Pionyr, and Bicara Therapeutics, and has served as a consultant or advisor for Merck, Novartis, Genentech/Roche, BMS, Compass Therapeutics, Rheos Medicines, Checkpoint Therapeutics, Bioentre, Gossamer Bio, Iovance Biotherapeutics, Catalym, Immunocore, Kairos Therapeutics, Zumutor Biologics, Corner Therapeutics, AstraZeneca, Curis, Pliant, Solu Therapeutics, Vir Biotechnology, and 92Bio, has received travel or expenses from Novartis and BMS, and holds several patents related to methods for treating MICA-related disorders, tumor antigens, immune checkpoint targets, and therapeutic peptides unrelated to this work. S.Sig. reports receiving commercial research grants from Bristol-Myers Squibb, AstraZeneca, Exelixis and Novartis. VAB has patents on the PD-1 pathway licensed by Bristol-Myers Squibb, Roche, Merck, EMD-Serono, Boehringer Ingelheim, AstraZeneca, Novartis and Dako unrelated to this work. A.K.S. reports compensation for consulting and/or scientific advisory board membership from Honeycomb Biotechnologies, Cellarity, Ochre Bio, Relation Therapeutics, Fog Pharma, Passkey Therapeutics, IntrECate Biotherapeutics, Bio-Rad Laboratories, and Dahlia Biosciences unrelated to this work. The other authors declare no competing interests.
Figures




References
-
- Marx Vivien. Method of the year: spatially resolved transcriptomics. Nature methods, 18 (1):9–14, 2021. - PubMed
-
- Nature Methods. Method of the year 2024: spatial proteomics. nature methods, 21:2195–2196, 2024. - PubMed
-
- Bodenmiller Bernd. Highly multiplexed imaging in the omics era: understanding tissue structures in health and disease. Nature Methods, 21(12):2209–2211, 2024. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials