

Chronic nicotine exposure induces molecular and transcriptomic endophenotypes associated with mood and anxiety disorders in a cerebral organoid neurodevelopmental model
- PMID: 39764466
- PMCID: PMC11701148
- DOI: 10.3389/fphar.2024.1473213
Chronic nicotine exposure induces molecular and transcriptomic endophenotypes associated with mood and anxiety disorders in a cerebral organoid neurodevelopmental model
Abstract
Introduction: Prenatal nicotine exposure (PNE) from maternal smoking disrupts regulatory processes vital to fetal development. These changes result in long-term behavioral impairments, including mood and anxiety disorders, that manifest later in life. However, the relationship underlying PNE, and the underpinnings of mood and anxiety molecular and transcriptomic phenotypes remains elusive.
Methods: To model nicotine exposure during prenatal development, our study used human cerebral organoids that were chronically exposed to nicotine and collected for molecular analyses.
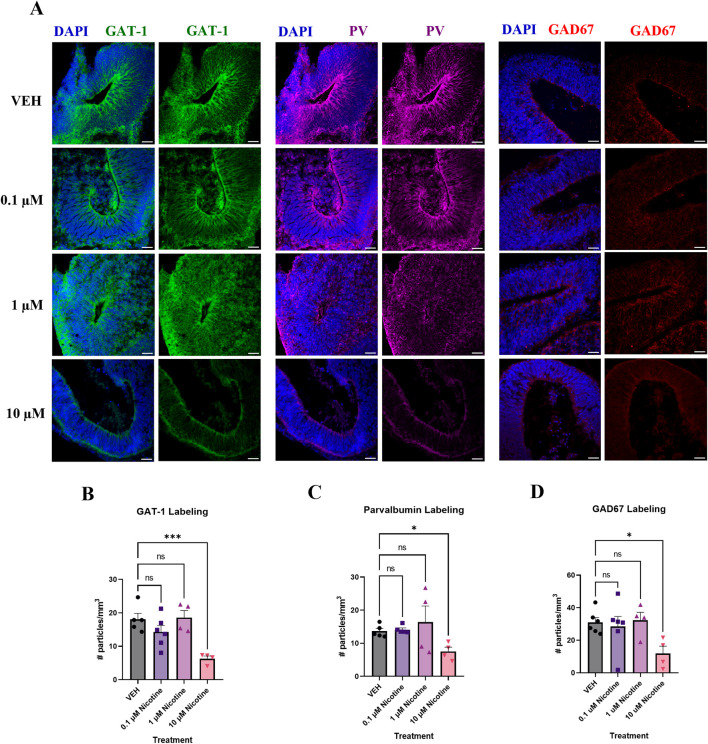
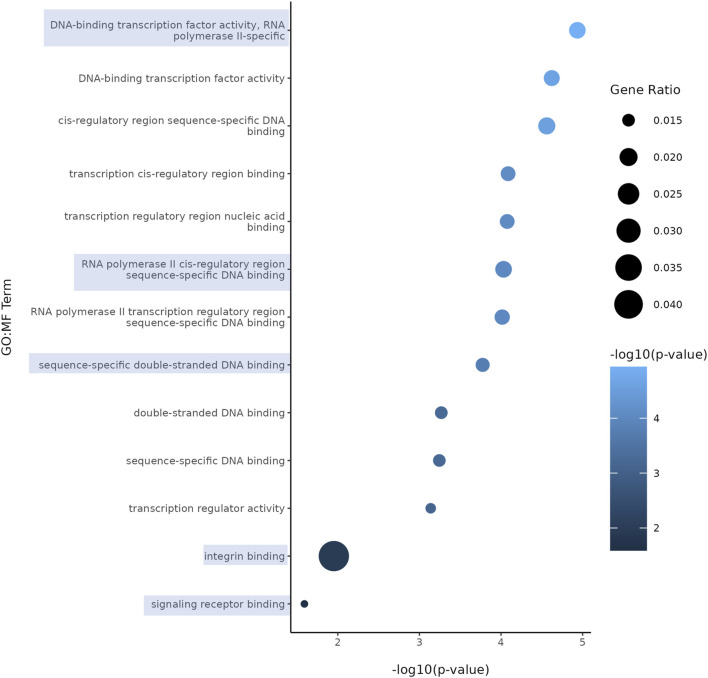
Results: Short-term, nicotine altered molecular markers of neural identity, mood and anxiety disorders and those involved in maintaining the excitatory/inhibitory (E/I) balance in the cortex. RNA sequencing further revealed transcriptomic changes in genes pertaining to embryonic development, neurogenesis, and DNA binding. Long-term, mature organoids demonstrated similar disruptions in E/I balance, decreased expression of neural identity markers, and altered dopamine receptor expression.
Discussion: Collectively, our results demonstrate that nicotine-induced alterations occur acutely and persist at later stages of development. These findings validate an in vitro model of PNE to better comprehend the emergence of neuropsychiatric molecular and transcriptomic endophenotypes resulting from gestational nicotine exposure.
Keywords: anxiety; depression; human cerebral organoids; prenatal development; prenatal nicotine exposure.
Copyright © 2024 Proud, Rodríguez-Ruiz, Gummerson, Vanin, Hardy, Rushlow and Laviolette.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Figures










References
-
- Alkam T., Nabeshima T. (2019). Prenatal nicotine exposure and neuronal progenitor cells. Neurosci. Nicotine Mech. Treat., 41–48. 10.1016/b978-0-12-813035-3.00006-x - DOI
-
- Aoyama Y., Toriumi K., Mouri A., Hattori T., Ueda E., Shimato A., et al. (2016). Prenatal nicotine exposure impairs the proliferation of neuronal progenitors, leading to fewer glutamatergic neurons in the medial prefrontal cortex. Neuropsychopharmacol 41, 578–589. 10.1038/npp.2015.186 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources