

Anchorage Accurately Assembles Anchor-Flanked Synthetic Long Reads
- PMID: 39764549
- PMCID: PMC11702288
- DOI: 10.4230/LIPIcs.WABI.2024.22
Anchorage Accurately Assembles Anchor-Flanked Synthetic Long Reads
Abstract
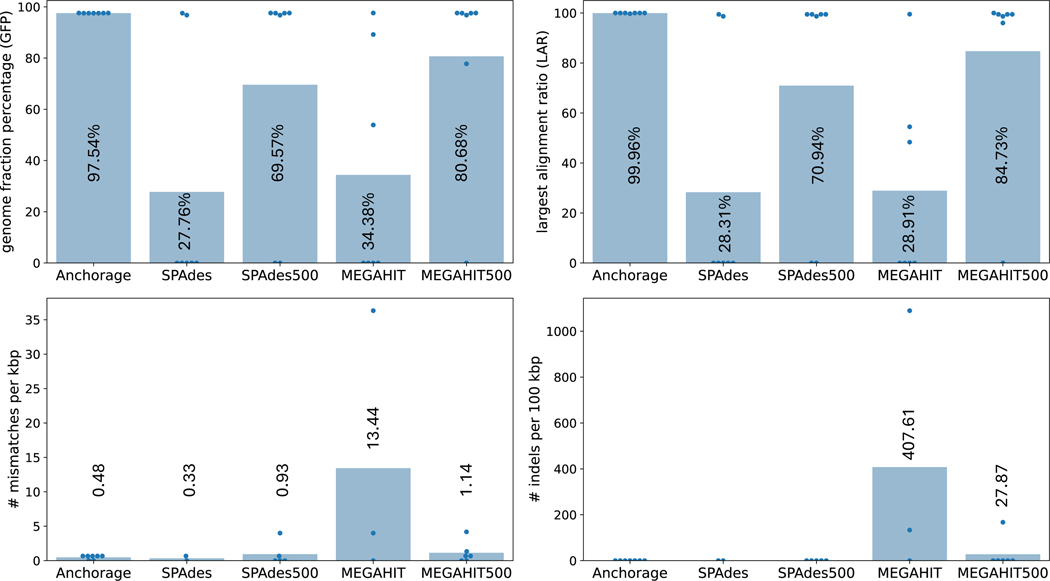
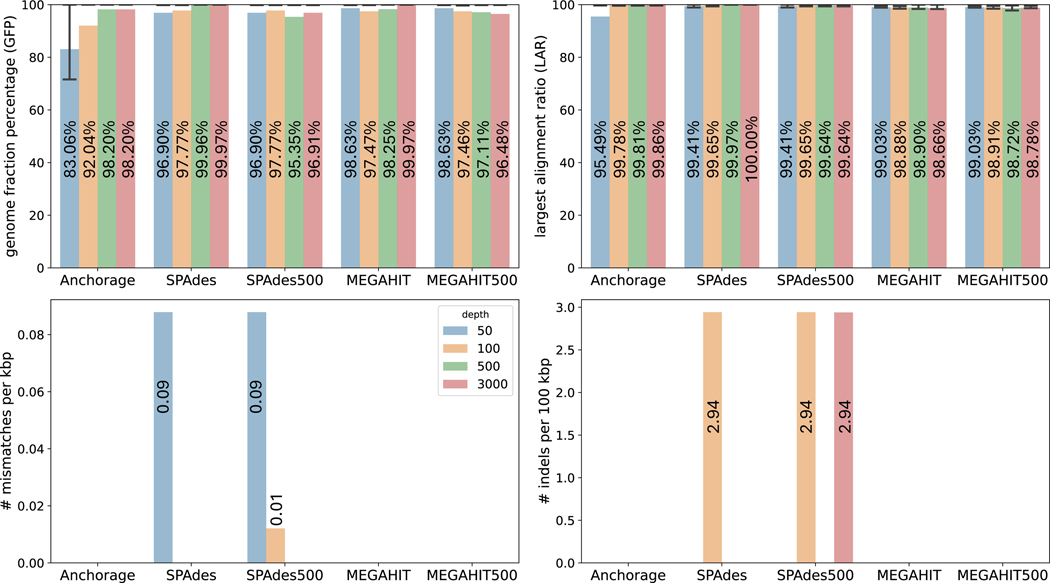
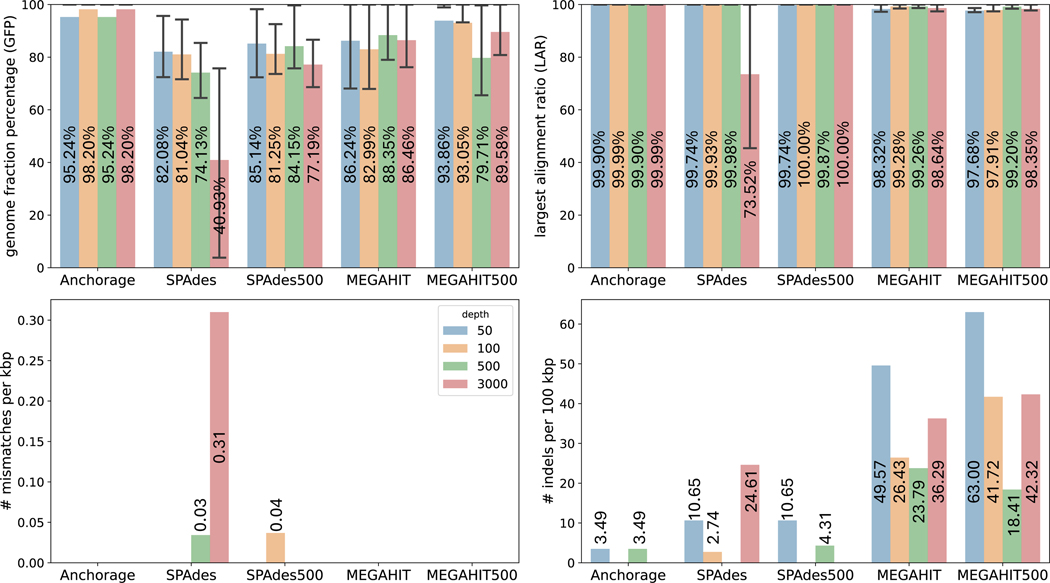
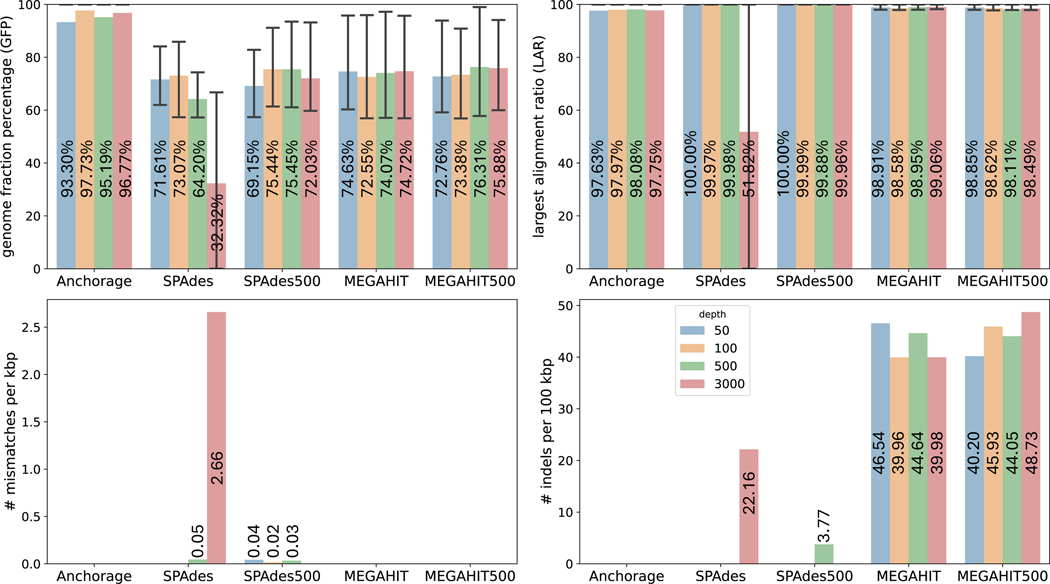
Modern sequencing technologies allow for the addition of short-sequence tags, known as anchors, to both ends of a captured molecule. Anchors are useful in assembling the full-length sequence of a captured molecule as they can be used to accurately determine the endpoints. One representative of such anchor-enabled technology is LoopSeq Solo, a synthetic long read (SLR) sequencing protocol. LoopSeq Solo also achieves ultra-high sequencing depth and high purity of short reads covering the entire captured molecule. Despite the availability of many assembly methods, constructing full-length sequence from these anchor-enabled, ultra-high coverage sequencing data remains challenging due to the complexity of the underlying assembly graphs and the lack of specific algorithms leveraging anchors. We present Anchorage, a novel assembler that performs anchor-guided assembly for ultra-high-depth sequencing data. Anchorage starts with a kmer-based approach for precise estimation of molecule lengths. It then formulates the assembly problem as finding an optimal path that connects the two nodes determined by anchors in the underlying compact de Bruijn graph. The optimality is defined as maximizing the weight of the smallest node while matching the estimated sequence length. Anchorage uses a modified dynamic programming algorithm to efficiently find the optimal path. Through both simulations and real data, we show that Anchorage outperforms existing assembly methods, particularly in the presence of sequencing artifacts. Anchorage fills the gap in assembling anchor-enabled data. We anticipate its broad use as anchor-enabled sequencing technologies become prevalent. Anchorage is freely available at https://github.com/Shao-Group/anchorage; the scripts and documents that can reproduce all experiments in this manuscript are available at https://github.com/Shao-Group/anchorage-test.
Keywords: Applied computing → Molecular sequence analysis; Genome assembly; LoopSeq; anchor-guided assembly; de Bruijn graph; synthetic long reads.
Conflict of interest statement
6Conflict of interest K.M., T.B.-Y., and R.K. are current or former employees of Element Biosciences and may hold stock options in the company.
Figures
References
-
- Bankevich Anton, Nurk Sergey, Antipov Dmitry, Gurevich Alexey A., Dvorkin Mikhail, Kulikov Alexander S., Lesin Valery M., Nikolenko Sergey I., Pham Son, Prjibelski Andrey D., Pyshkin Alexey V., Sirotkin Alexander V., Vyahhi Nikolay, Tesler Glenn, Alekseyev Max A., and Pevzner Pavel A. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. Journal of Computational Biology, 19(5):455–477, 2012. - PMC - PubMed
-
- Benton Briana, King Stephen, Greenfield Samuel R., Puthuveetil Nikhita, Reese Amy L., Duncan James, Marlow Robert, Tabron Corina, Pierola Amanda E., Yarmosh David A., Combs Patrick Ford, Riojas Marco A., Bagnoli John, and Jacobs Jonathan L. The ATCC Genome Portal: Microbial Genome Reference Standards with Data Provenance. Microbiology Resource Announcements, 10(47):e00818–21, 2023. - PMC - PubMed
-
- Birol Inanç, Jackman Shaun D., Nielsen Cydney B., Qian Jenny Q., Varhol Richard, Stazyk Greg, Morin Ryan D., Zhao Yongjun, Hirst Martin, Schein Jacqueline E., Horsman Doug E., Connors Joseph M., Gascoyne Randy D., Marra Marco A., and Jones Steven J. M. De novo transcriptome assembly with ABySS. Bioinformatics, 25(21):2872–2877, 2009. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous