

Side- and Disease-Dependent Changes in Human Aortic Valve Cell Population and Transcriptomic Heterogeneity Determined by Single-Cell RNA Sequencing
- PMID: 39766890
- PMCID: PMC11675841
- DOI: 10.3390/genes15121623
Side- and Disease-Dependent Changes in Human Aortic Valve Cell Population and Transcriptomic Heterogeneity Determined by Single-Cell RNA Sequencing
Abstract
Background: Calcific aortic valve disease (CAVD) is a highly prevalent disease, especially in the elderly population, but there are no effective drug therapies other than aortic valve repair or replacement. CAVD develops preferentially on the fibrosa side, while the ventricularis side remains relatively spared through unknown mechanisms. We hypothesized that the fibrosa is prone to the disease due to side-dependent differences in transcriptomic patterns and cell phenotypes.
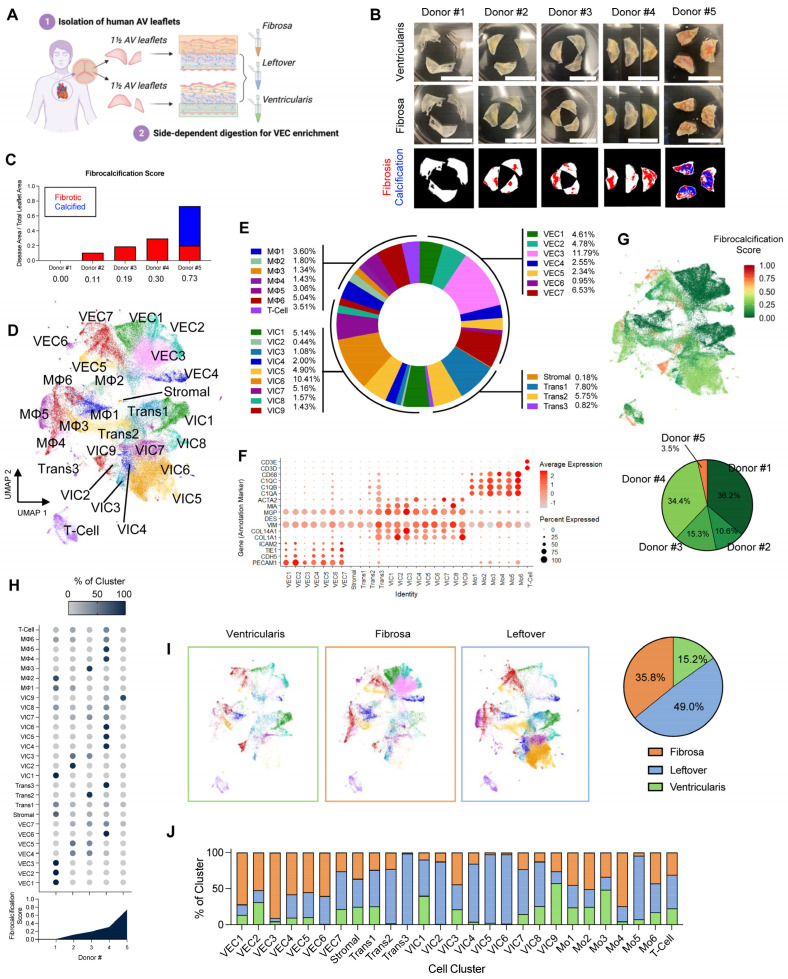
Methods: To test this hypothesis, we performed single-cell RNA sequencing using a new method to collect endothelial-enriched samples independently from the fibrosa and ventricularis sides of freshly obtained human aortic valve leaflets from five donors, ranging from non-diseased to fibrocalcific stages.
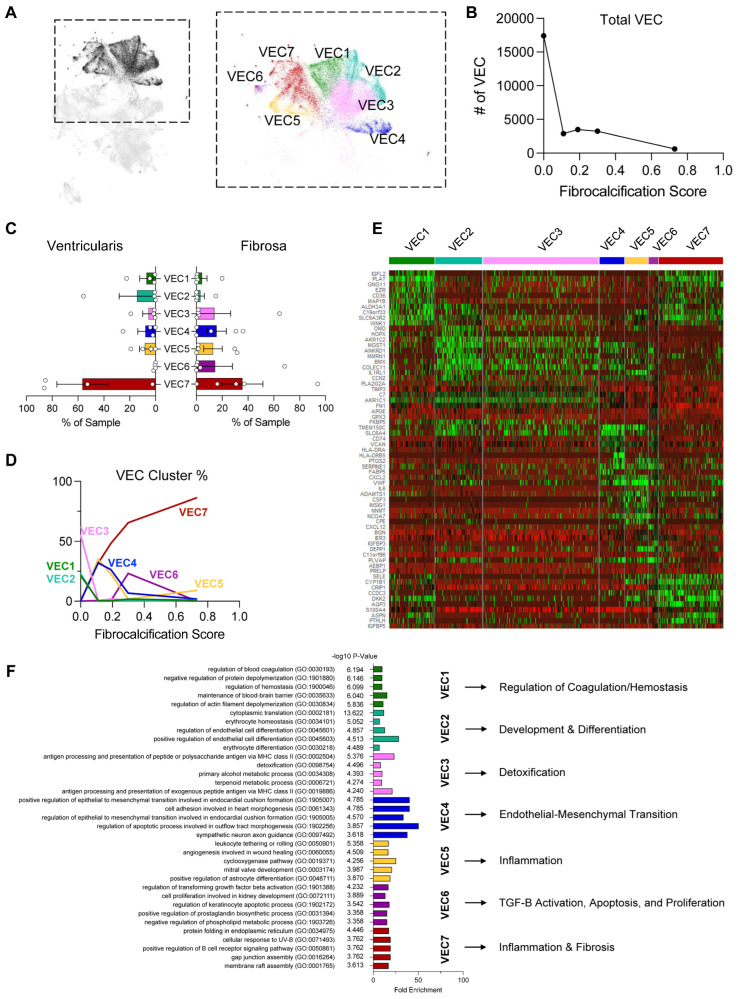
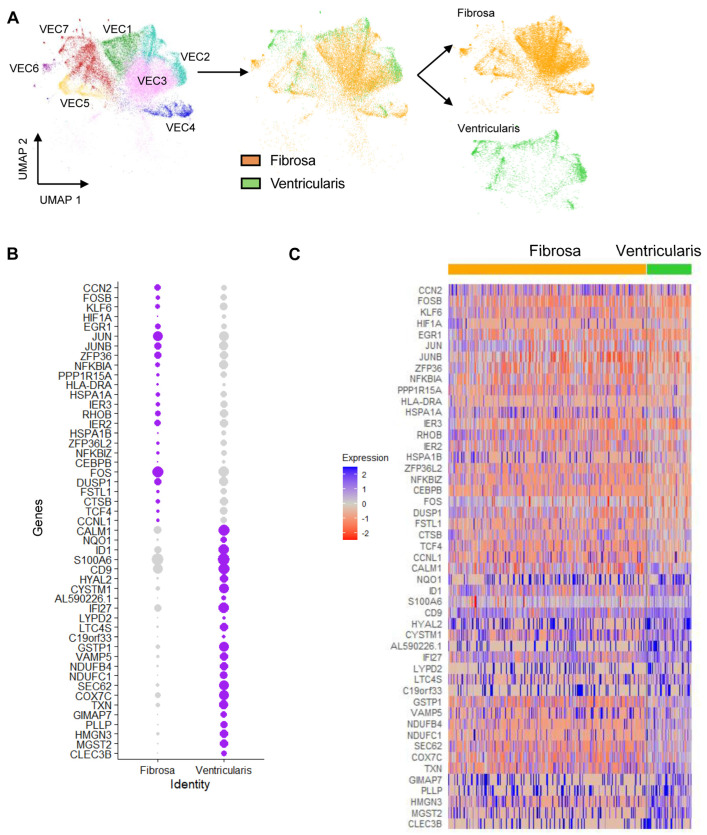
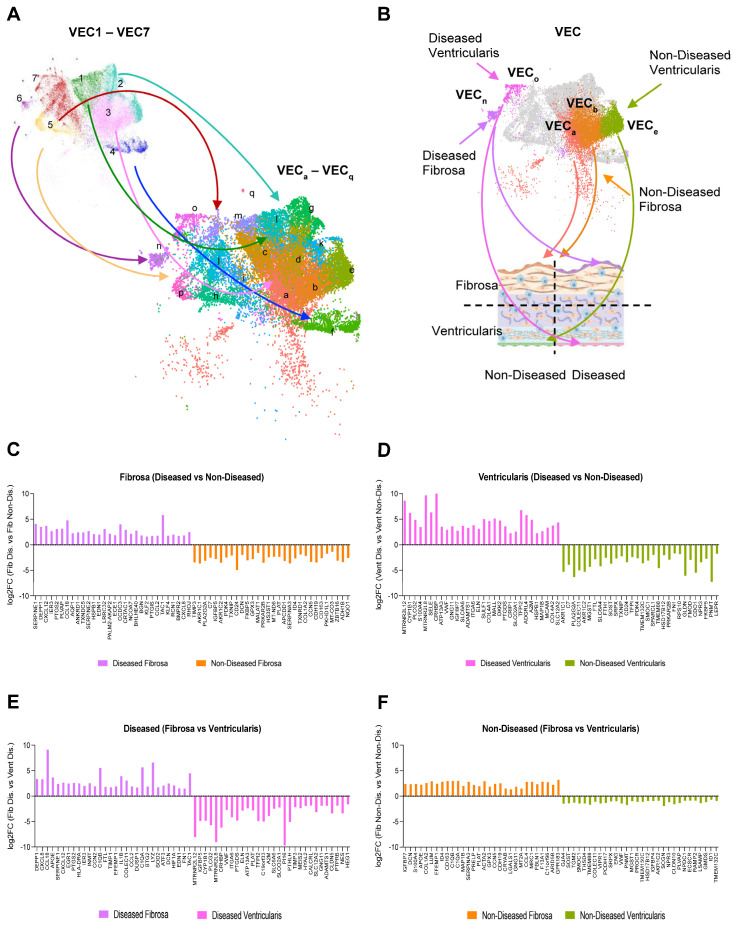
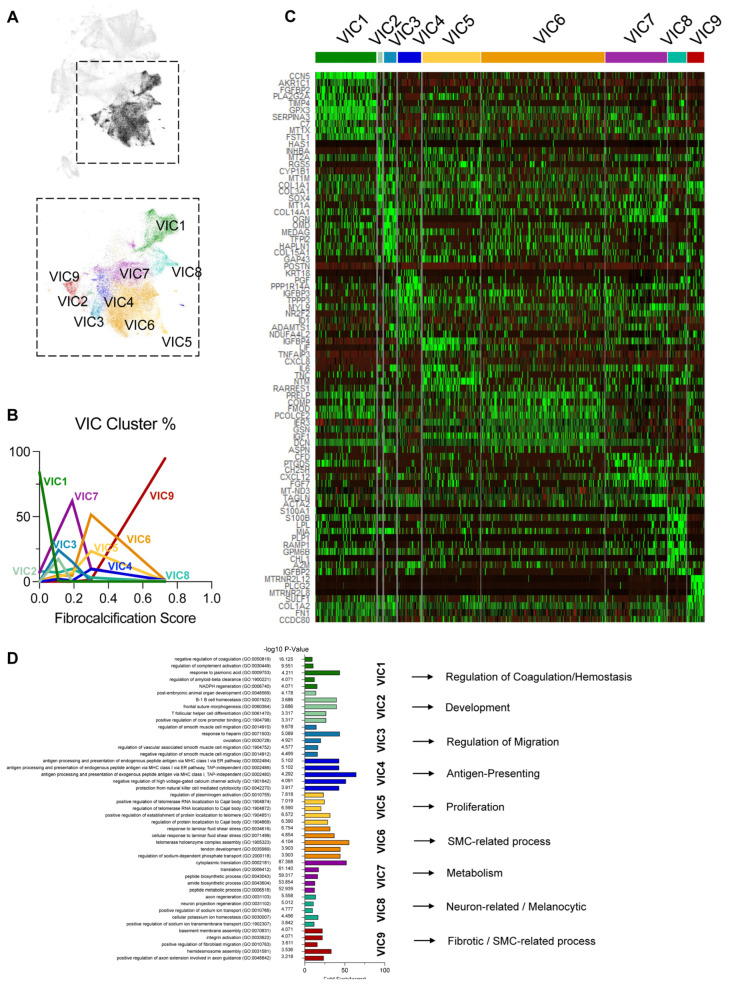
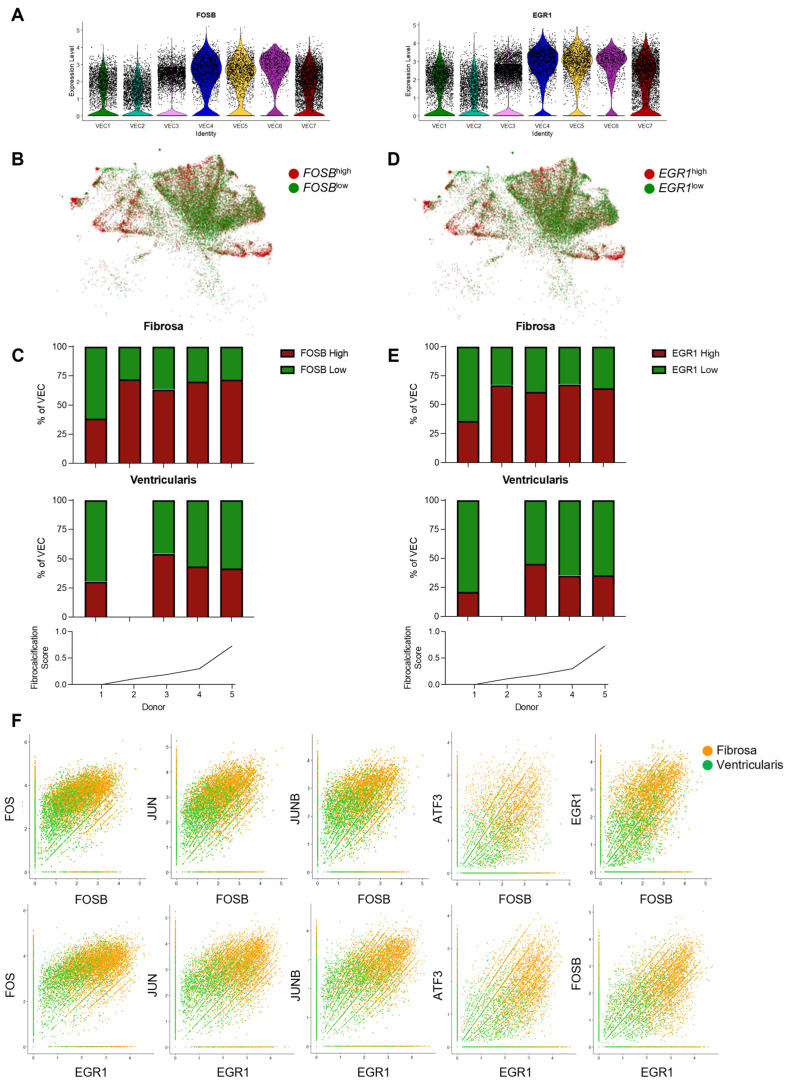
Results: From the 82,356 aortic valve cells analyzed, we found 27 cell clusters, including seven valvular endothelial cell (VEC), nine valvular interstitial cell (VIC), and seven immune, three transitional, and one stromal cell population. We identified several side-dependent VEC subtypes with unique gene expression patterns. Homeostatic VIC clusters were abundant in non-diseased tissues, while VICs enriched with fibrocalcific genes and pathways were more prevalent in diseased leaflets. Furthermore, homeostatic macrophage (MΦ) clusters decreased while inflammatory MΦ and T-cell clusters increased with disease progression. A foamy MΦ cluster was increased in the fibrosa of mildly diseased tissues. Some side-dependent VEC clusters represented non-diseased, protective phenotypes, while others were CAVD-associated and were characterized by genes enriched in pathways of inflammation, endothelial-mesenchymal transition, apoptosis, proliferation, and fibrosis. Interestingly, we found several activator protein-1 (AP-1)-related transcription factors (FOSB, FOS, JUN, JUNB) and EGR1 to be upregulated in the fibrosa and diseased aortic valve leaflets.
Conclusions: Our results showed that VECs are highly heterogeneous in a side- and CAVD-dependent manner. Unique VEC clusters and their differentially regulated genes and pathways found in the fibrosa of diseased tissues may represent novel pathogenic mechanisms and potential therapeutic targets.
Keywords: AP-1 related transcription factors; aortic sclerosis; aortid stenosis; calcific aortic valve disease; endothelial-to-mesenchymal transition; human aortic valves; inflammation; single-cell RNA sequencing; transcriptomics; valve endothelial cells; valve interstitial cells.
Conflict of interest statement
H.J. is the founder of Flokines Pharma. All other authors report no conflicts of interest.
Figures
References
-
- Virani S.S., Alonso A., Aparicio H.J., Benjamin E.J., Bittencourt M.S., Callaway C.W., Carson A.P., Chamberlain A.M., Cheng S., Delling F.N., et al. Heart disease and stroke statistics-2021 update: A report from the american heart association. Circulation. 2021;143:e254–e743. - PubMed
-
- Yadgir S., Johnson C.O., Aboyans V., Adebayo O.M., Adedoyin R.A., Afarideh M., Alahdab F., Alashi A., Alipour V., Arabloo J., et al. Global, regional, and national burden of calcific aortic valve and degenerative mitral valve diseases, 1990–2017. Circulation. 2020;141:1670–1680. doi: 10.1161/CIRCULATIONAHA.119.043391. - DOI - PubMed
-
- Otto C.M., Nishimura R.A., Bonow R.O., Carabello B.A., Erwin J.P., 3rd, Gentile F., Jneid H., Krieger E.V., Mack M., McLeod C., et al. 2020 ACC/AHA guideline for the management of patients with valvular heart disease: A report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2021;143:e72–e227. - PubMed
MeSH terms
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
