

Phenotypic and Genetic Heterogeneity of a Pakistani Cohort of 15 Consanguineous Families Segregating Variants in Leber Congenital Amaurosis-Associated Genes
- PMID: 39766915
- PMCID: PMC11728111
- DOI: 10.3390/genes15121646
Phenotypic and Genetic Heterogeneity of a Pakistani Cohort of 15 Consanguineous Families Segregating Variants in Leber Congenital Amaurosis-Associated Genes
Abstract
Background: Leber congenital amaurosis (LCA) is a congenital onset severe form of inherited retinal dystrophy (IRD) and a common cause of pediatric blindness. Disease-causing variants in at least 14 genes are reported to predispose LCA phenotype. LCA is inherited as an autosomal recessive disease. It can be an isolated eye disorder or as part of a syndrome, such as Senior Loken or Joubert syndrome. Sequencing studies from consanguineous populations have proven useful for novel variants identification; thus, the present study aimed to explore the genetic heterogeneity of 15 consanguineous Pakistani families, each segregating a severe IRD phenotype using targeted next generation sequencing.
Methods: This study enrolled 15 consanguineous families, each with multiple affected cases of retinal dystrophy phenotype. DNA was extracted from blood samples. Targeted panel sequencing of 344 known genes for IRDs was performed, followed by Sanger sequencing for segregation analysis.
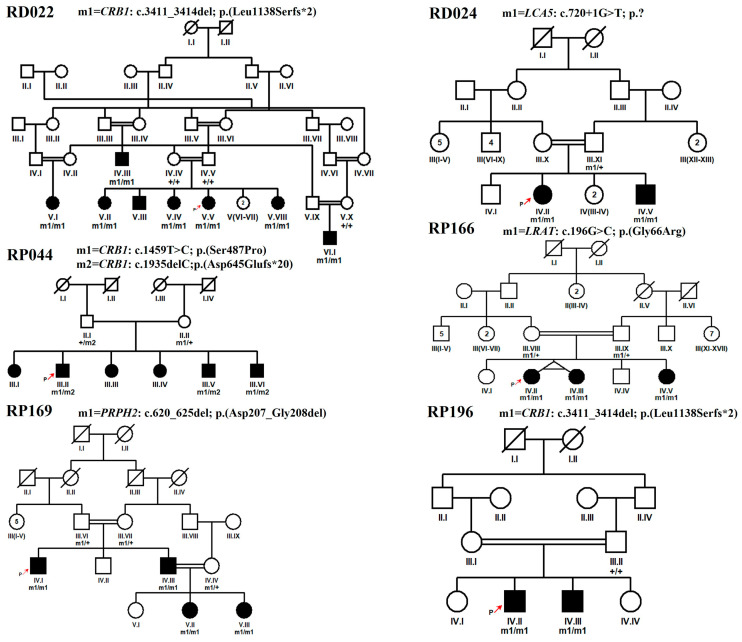
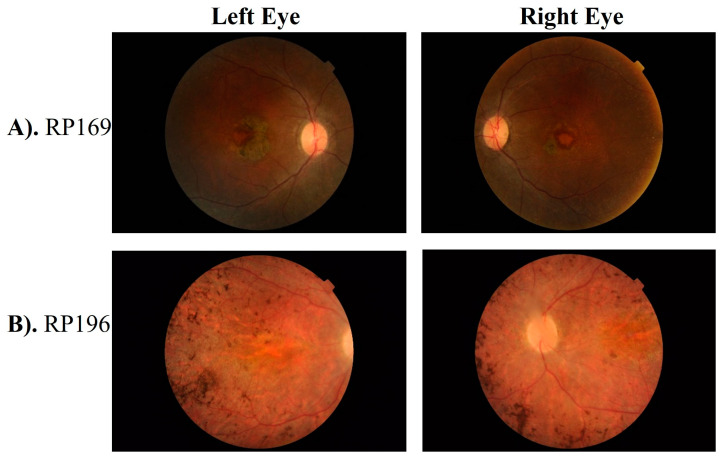
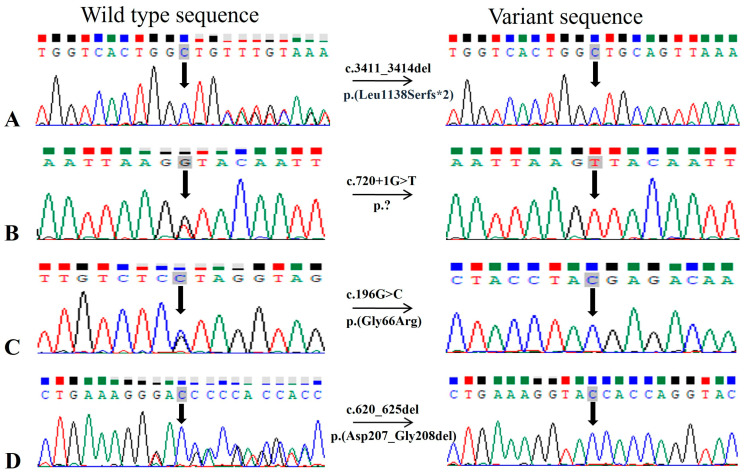
Results: Data analysis revealed a total of eight reported (c.316C>T and c.506G>A in RDH12; c.864dup and c.1012C>T in SPATA7, as well as c.1459T>C, c.1062_1068del, c.1495+1G>A, c.998G>A in the CRB1, LCA5, TULP1, and IFT140 genes, respectively) and four novel homozygous (c.720+1G>T in LCA5, c.196G>C in LRAT, c.620_625del in PRPH2, and c.3411_3414del in CRB1) variants segregating with disease phenotype in each respective family. Furthermore, a novel heterozygous variant of CRB1 gene, i.e., c.1935delC in compound heterozygous condition was found segregating with disease phenotype in one large family with multiple consanguinity loops.
Conclusion: Comprehensive molecular diagnosis of 15 consanguineous Pakistani families led to the identification of a total of 5 novel variants contributing to genetic heterogeneity of LCA-associated genes and helped to provide genetic counseling to the affected families.
Keywords: Leber congenital amaurosis; autosomal recessive; childhood blindness; genetic heterogeneity.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
Similar articles
-
Genetic and Clinical Profile of Retinopathies Due to Disease-Causing Variants in Leber Congenital Amaurosis (LCA)-Associated Genes in a Large German Cohort.Int J Mol Sci. 2023 May 17;24(10):8915. doi: 10.3390/ijms24108915. Int J Mol Sci. 2023. PMID: 37240262 Free PMC article.
-
Screening of a large cohort of leber congenital amaurosis and retinitis pigmentosa patients identifies novel LCA5 mutations and new genotype-phenotype correlations.Hum Mutat. 2013 Nov;34(11):1537-1546. doi: 10.1002/humu.22398. Epub 2013 Sep 17. Hum Mutat. 2013. PMID: 23946133 Free PMC article.
-
Deciphering the Genetic Basis of Degenerative and Developmental Eye Disorders in 50 Pakistani Consanguineous Families Using Whole-Exome Sequencing.Int J Mol Sci. 2025 Mar 18;26(6):2715. doi: 10.3390/ijms26062715. Int J Mol Sci. 2025. PMID: 40141357 Free PMC article.
-
CRB1-Related Leber Congenital Amaurosis: Reporting Novel Pathogenic Variants and a Brief Review on Mutations Spectrum.Iran Biomed J. 2019 Sep;23(5):362-8. doi: 10.29252/.23.5.362. Epub 2019 May 19. Iran Biomed J. 2019. PMID: 31103025 Free PMC article. Review.
-
Leber Congenital Amaurosis – RETIRED CHAPTER, FOR HISTORICAL REFERENCE ONLY.2004 Jul 7 [updated 2013 May 2]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. 2004 Jul 7 [updated 2013 May 2]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. PMID: 20301475 Free Books & Documents. Review.
References
-
- Hosono K., Nishina S., Yokoi T., Katagiri S., Saitsu H., Kurata K., Miyamichi D., Hikoya A., Mizobuchi K., Nakano T., et al. Molecular Diagnosis of 34 Japanese Families with Leber Congenital Amaurosis Using Targeted Next Generation Sequencing. Sci. Rep. 2018;8:8279. doi: 10.1038/s41598-018-26524-z. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
