

The Causal Role of Ectopic Fat Deposition in the Pathogenesis of Metabolic Syndrome
- PMID: 39769002
- PMCID: PMC11675790
- DOI: 10.3390/ijms252413238
The Causal Role of Ectopic Fat Deposition in the Pathogenesis of Metabolic Syndrome
Abstract
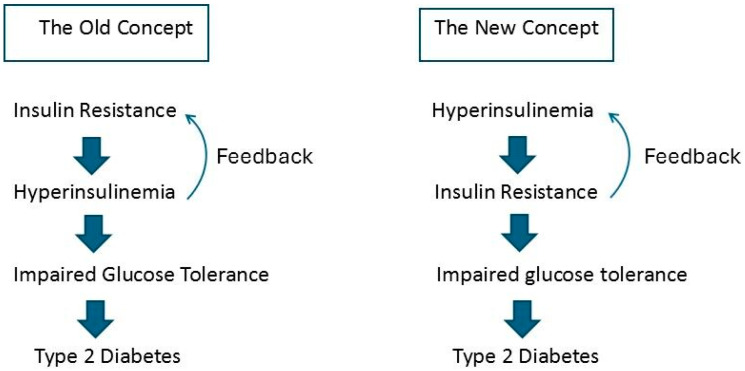
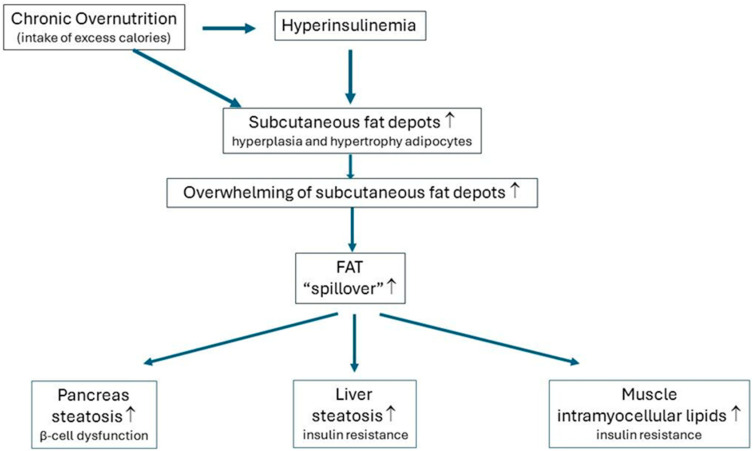
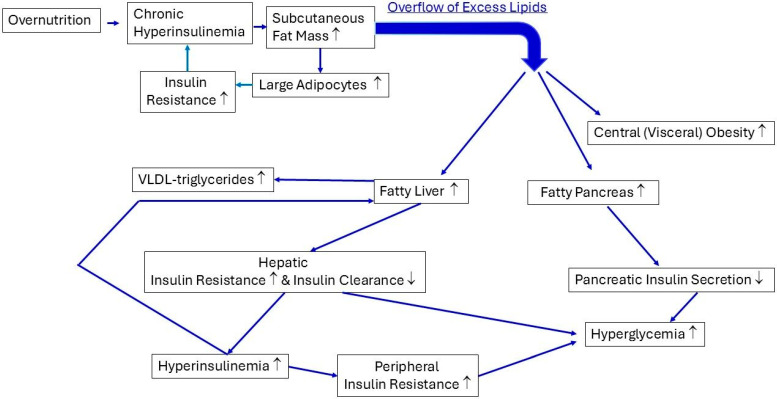
Consuming a "modern" Western diet and overnutrition may increase insulin secretion. Additionally, nutrition-mediated hyperinsulinemia is a major driver of ectopic fat deposition. The global prevalence of metabolic syndrome is high and growing. Within this context, people with congenital lipodystrophy often experience a severe form of metabolic syndrome. Evidence is increasingly supporting that subtle partial lipodystrophy plays an important role in the development of metabolic syndrome in the general population. In individuals in the general population with subtle partial lipodystrophy, as well as in those with congenital lipodystrophy, the subcutaneous adipose tissues are unable to accommodate surplus energy intake. In both conditions, (excess) fat is directed toward the liver, pancreas, and muscles, where it is deposited as ectopic fat, as this fat can no longer be stored in the "safe" subcutaneous fat depots. Ectopic fat depositions cause insulin resistance in the liver and muscles, as well as β-cell dysfunction in the pancreas. Support of a direct pathological role of ectopic fat deposition in this condition is further provided by the rapid normalization of hepatic insulin sensitivity and improvement in pancreatic β-cell function after marked reductions in ectopic fat depositions. Thus, ectopic fat deposition in the liver, pancreas, and muscles may play a causal role in the pathogenesis of metabolic syndrome even in the general population. As such, the prevention of ectopic fat deposition may reduce the risk of metabolic syndrome and mitigate its effects.
Keywords: abdominal (visceral) obesity; ectopic fat deposition; hyperinsulinemia; insulin resistance; lipodystrophy; metabolic syndrome; overnutrition; type 2 diabetes.
Conflict of interest statement
The author declares no conflicts of interest.
Figures
References
-
- Vague J. La différenciation sexuelle; facteur déterminant des formes de l’obésité [Sexual differentiation; Factor determining forms of obesity] Presse Med. 1947;55:339. - PubMed
-
- Vague J. Significance of obesity in medical practice. Mars Med. 1953;90:179–189. - PubMed
-
- Carey D.G., Jenkins A.B., Campbell L.V., Freund J., Chisholm D.J. Abdominal fat and insulin resistance in normal and overweight women: Direct measurements reveal a strong relationship in subjects at both low and high risk of NIDDM. Diabetes. 1996;45:633–638. doi: 10.2337/diab.45.5.633. - DOI - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Research Materials
