

Practical Recommendations for the Diagnosis and Management of Lysosomal Acid Lipase Deficiency with a Focus on Wolman Disease
- PMID: 39770929
- PMCID: PMC11678757
- DOI: 10.3390/nu16244309
Practical Recommendations for the Diagnosis and Management of Lysosomal Acid Lipase Deficiency with a Focus on Wolman Disease
Abstract
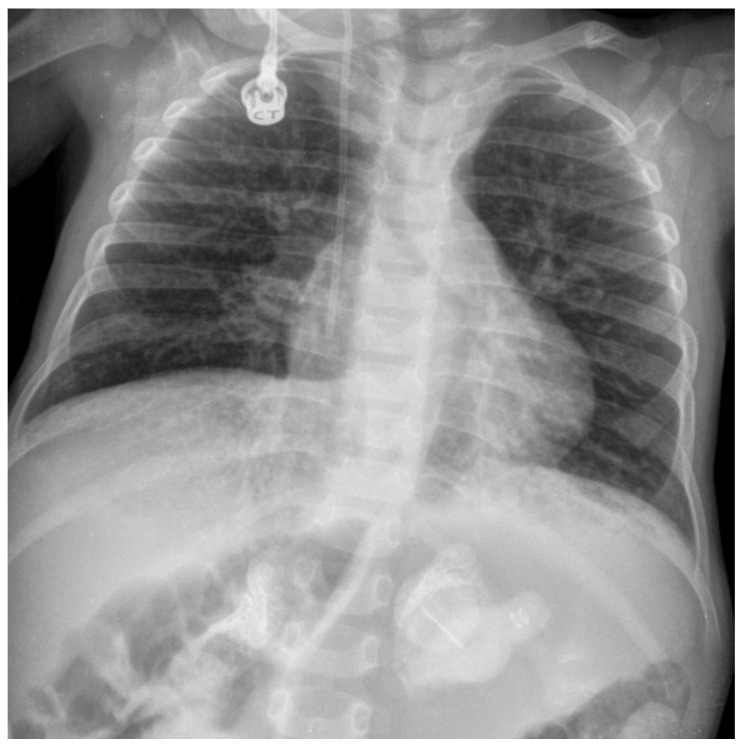
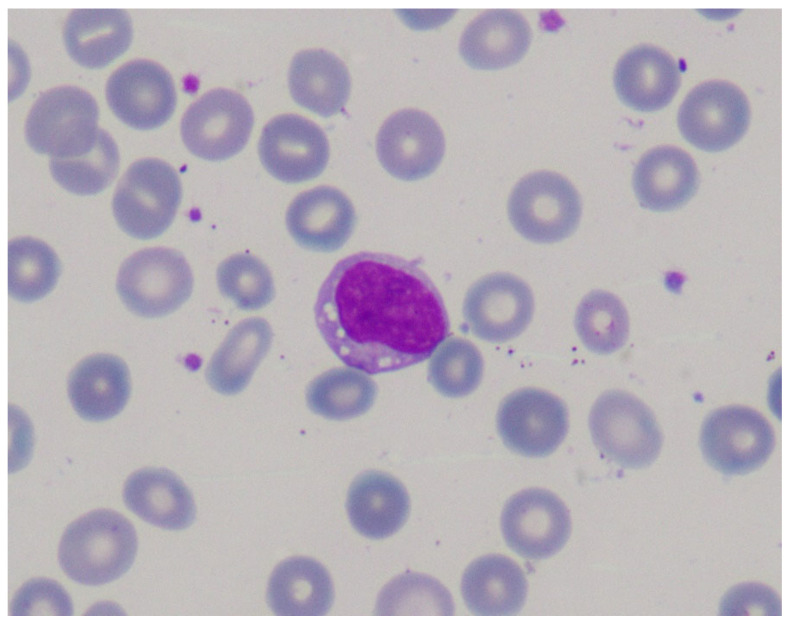
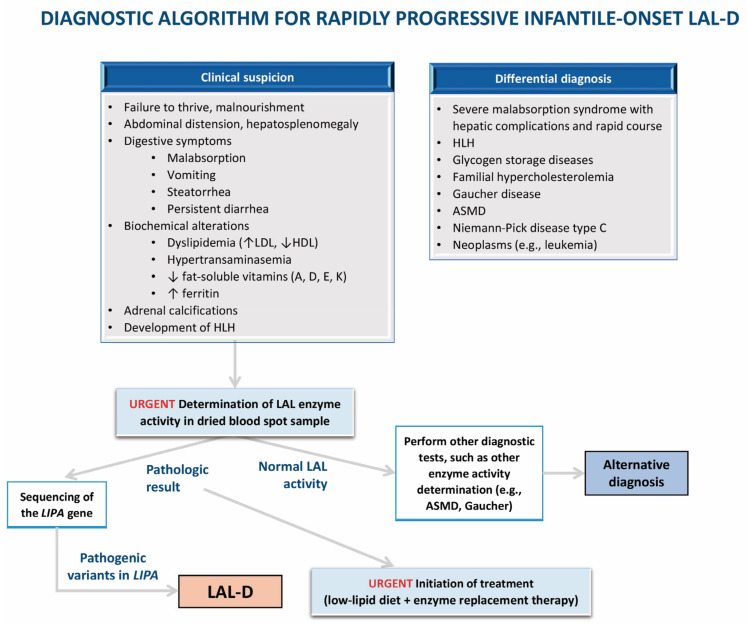
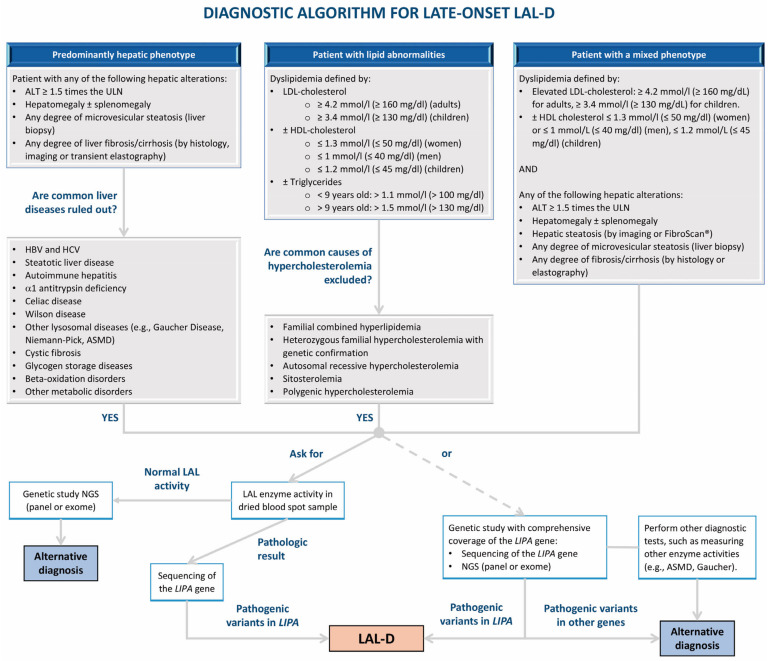
Lysosomal acid lipase deficiency (LAL-D) is an ultra-rare lysosomal storage disease with two distinct phenotypes, an infantile-onset form (formerly Wolman disease) and a later-onset form (formerly cholesteryl ester storage disease). The objective of this narrative review is to examine the most important aspects of the diagnosis and treatment of LAL-D and to provide practical expert recommendations. The infantile-onset form occurs in the first weeks of life and is characterized by malnourishment and failure to thrive due to gastrointestinal impairment (vomiting, diarrhea, malabsorption), as well as systemic inflammation, hepatosplenomegaly, and adrenal calcifications. Mortality is close to 100% before one year of life in the absence of specific treatment. The later-onset form can be diagnosed in childhood or adulthood and is characterized by chronic liver injury and/or lipid profile alterations. When LAL-D is suspected, enzyme activity should be determined to confirm the diagnosis, with analysis from a dried blood spot sample being the quickest and most reliable method. In infantile-onset LAL-D, the initiation of enzyme replacement therapy (sebelipase α) and careful nutritional management with a low-lipid diet is very urgent, as prognosis is directly linked to the early initiation of specific treatment. In recent years, our knowledge of the management of LAL-D has increased considerably, with improvements regarding the initial enzyme replacement therapy dose and careful nutritional treatment with a low-lipid diet to decrease lipid deposition and systemic inflammation, leading to better outcomes. In this narrative review we offer a quick guide for the initial management of infantile-onset LAL-D.
Keywords: LAL-D; LIPA gene; Wolman disease; cholesteryl ester storage disease; diet; enzyme replacement therapy; infantile-onset lysosomal acid lipase deficiency; lysosomal acid lipase; lysosomal acid lipase deficiency; sebelipase.
Conflict of interest statement
Javier de las Heras has received speaking and scientific consultancy fees from Astra Zeneca-Alexion. Carolina Almohalla received speaking fees from Astra Zeneca-Alexion. Javier Blasco-Alonso declares no conflicts of interest. Mafalda Bourbon received a project grant from SOBI. María-Luz Couce declares no conflicts of interest. María José de Castro López declares no conflicts of interest. Mª Concepción García Jiménez received speaking fees from Astra Zeneca-Alexion. David Gil Ortega received speaking fees from Astra Zeneca-Alexion. Luisa Gonzalez-Dieguez received speaking and scientific consultancy fees from Astra Zeneca-Alexion. Silvia Meavilla declares no conflicts of interest. Ana Moreno-Álvarez received speaking fees from Astra Zeneca-Alexion. José Pastor-Rosado has received conference payments from Astra Zeneca-Alexion; grants for attendance at scientific meetings, as well as fees for scientific consultancy from Astra Zeneca-Alexion Alexion. Paula Sánchez-Pintos received speaking fees from Astra Zeneca-Alexion. Irene Serrano-Gonzalo received speaking fees from Astra Zeneca-Alexion. Eduardo López is president of the Spanish LAL-D Patient Organization. That organization has received consultancy fees from Astra Zeneca-Alexion. Pedro Valdivielso has received honoraria for consulting, expert testimony, conferences, presentations, manuscripts or educational events from AstraZeneca-Alexion, Amgen, Sanofi, Novartis, Ferrer and Daiichi-Sankyo; meeting support from Amgen, Novartis, Ferrer, MSD, Daiichi-Sankyo, and Sanofi; and research grants/contracts from Ferrer, SOBI, Lipigon, Ionis, and Capenergy. Raquel Yahyaoui has received speaking fees and a research grant from Astra Zeneca-Alexion. Jesús Quintero has received honoraria for consulting, conferences, presentations, manuscripts or educational events from AstraZeneca-Alexion, IPSEN, Miru, and Orphalan; and research grants/contracts from IPSEN.
Figures
References
-
- Reiner Z., Guardamagna O., Nair D., Soran H., Hovingh K., Bertolini S., Jones S., Coric M., Calandra S., Hamilton J., et al. Lysosomal acid lipase deficiency: An under-recognized cause of dyslipidaemia and liver dysfunction. Atherosclerosis. 2014;235:21–30. - PubMed
-
- Bernstein D.L., Hulkova H., Bialer M.G., Desnick R.J. Cholesteryl ester storage disease: Review of the findings in 135 reported patients with an underdiagnosed disease. J. Hepatol. 2013;58:1230–1243. - PubMed
-
- Jones S.A., Valayannopoulos V., Schneider E., Eckert S., Banikazemi M., Bialer M., Cederbaum S., Chan A., Dhawan A., Di Rocco M., et al. Rapid progression and mortality of lysosomal acid lipase deficiency presenting in infants. Genet. Med. 2016;18:452–458. doi: 10.1038/gim.2015.108. - DOI - PMC - PubMed
-
- Balwani M., Balistreri W., D’Antiga L., Evans J., Ros E., Abel F., Wilson D.P. Lysosomal acid lipase deficiency manifestations in children and adults: Baseline data from an international registry. Liver Int. 2023;43:1537–1547. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
