

Investigation of RNA Viruses in Culicoides Latreille, 1809 (Diptera: Ceratopogonidae) in a Mining Complex in the Southeastern Region of the Brazilian Amazon
- PMID: 39772171
- PMCID: PMC11728802
- DOI: 10.3390/v16121862
Investigation of RNA Viruses in Culicoides Latreille, 1809 (Diptera: Ceratopogonidae) in a Mining Complex in the Southeastern Region of the Brazilian Amazon
Abstract
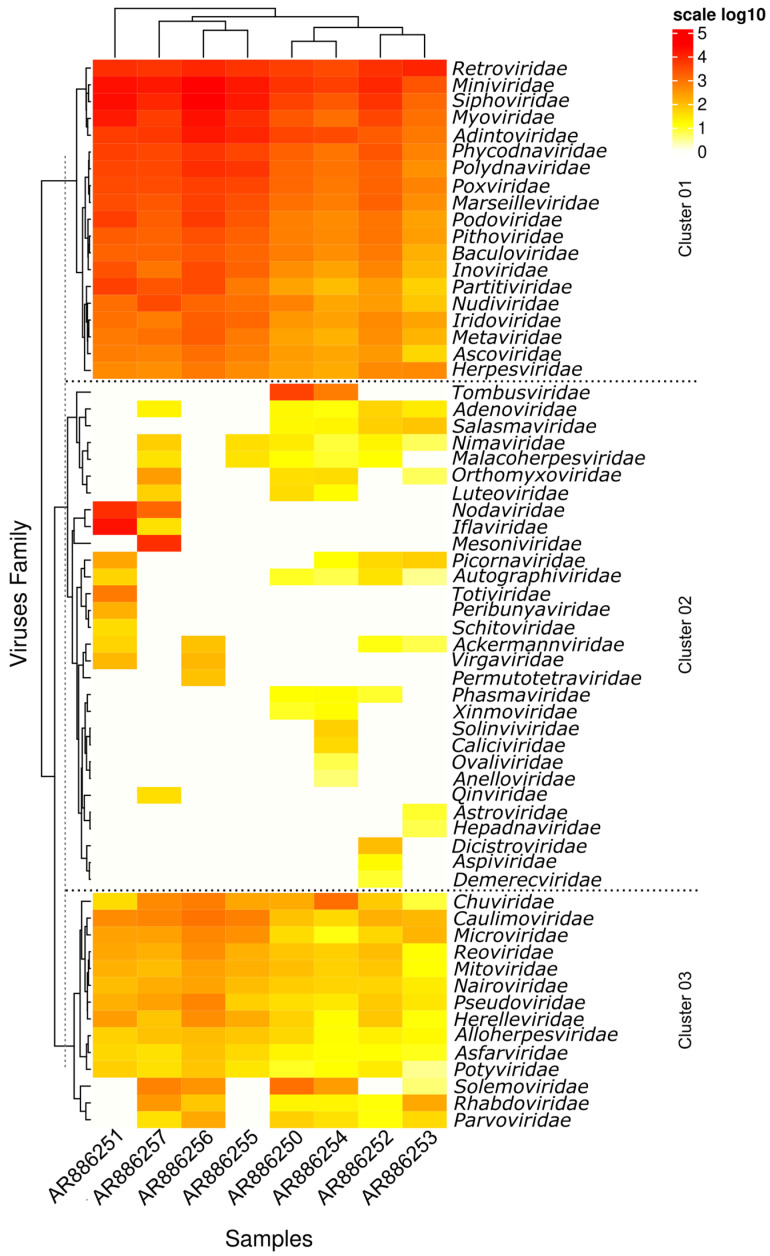
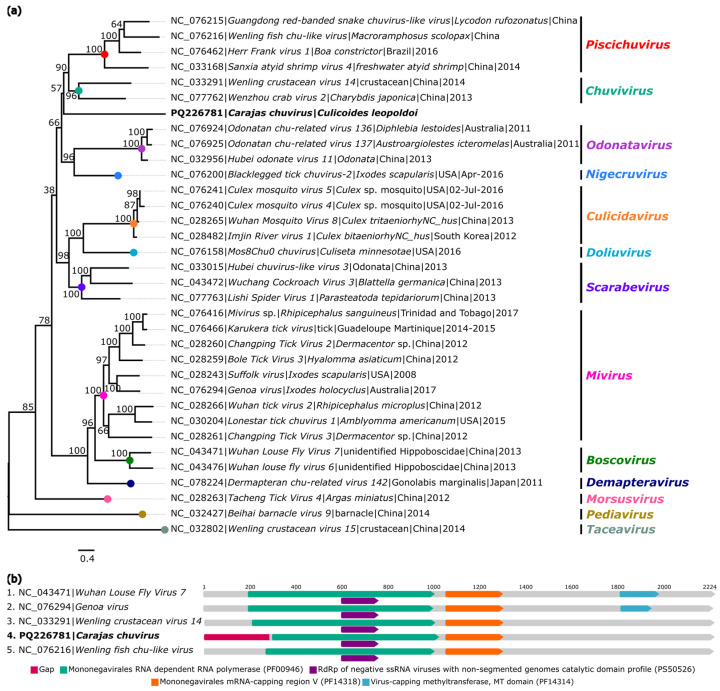
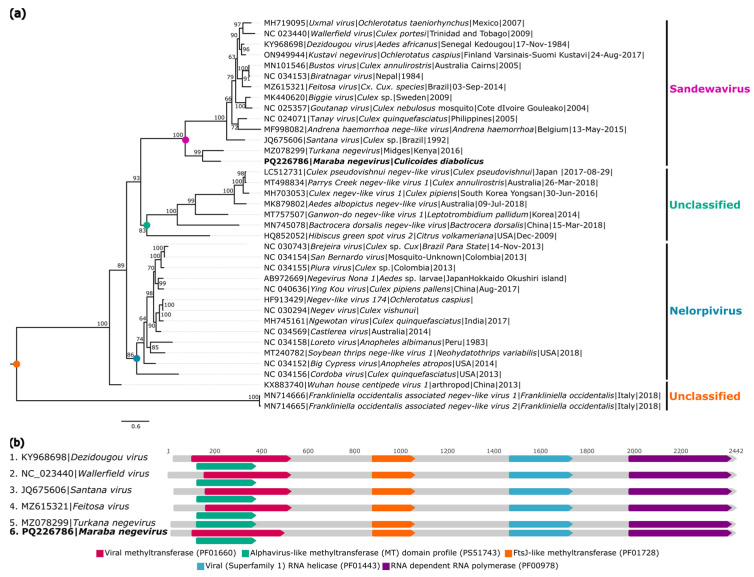
The biting midges Culicoides Latreille, 1809 (Diptera: Ceratopogonidae) is highly relevant to epidemiology and public health, as it includes species that are potential vectors of human and animal arboviruses. The aim of this study was to investigate the presence of RNA viruses in species of the genus Culicoides collected in the Carajás mining complex in the state of Pará. The biting midges were collected in the municipalities of Canaã dos Carajás, Curionópolis and Marabá and morphologically identified. A total of 1139 specimens of seven Culicoides species were grouped into eight pools and subjected to metagenomic analysis. Eight new insect-specific viruses (ISVs) were characterized and assigned to the order Tolivirales, the families Chuviridae, Nodaviridae, Iflaviridae, Mesoniviridae, and Flaviviridae, and the taxon Negevirus. All viruses identified were assigned to clades, families and taxa never reported in Culicoides in Brazil. This study demonstrated that biting midges harbor a significant diversity of RNA viruses, many of which are still unknown, highlighting the importance of studies aiming at virome of these insects.
Keywords: Culicoides; biting midges; insect specific-viruses; metagenomic; virome; viruses.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures






Similar articles
-
Viral metagenomics of hematophagous insects collected in the Carajas mining complex, Pará State, Brazil.Acta Trop. 2025 Mar;263:107551. doi: 10.1016/j.actatropica.2025.107551. Epub 2025 Feb 10. Acta Trop. 2025. PMID: 39938727
-
Profiling of RNA Viruses in Biting Midges (Ceratopogonidae) and Related Diptera from Kenya Using Metagenomics and Metabarcoding Analysis.mSphere. 2021 Oct 27;6(5):e0055121. doi: 10.1128/mSphere.00551-21. Epub 2021 Oct 13. mSphere. 2021. PMID: 34643419 Free PMC article.
-
Characterisation of the virome of Culicoides brevitarsis Kieffer (Diptera: Ceratopogonidae), a vector of bluetongue virus in Australia.J Gen Virol. 2025 Feb;106(2):002076. doi: 10.1099/jgv.0.002076. J Gen Virol. 2025. PMID: 39976626 Free PMC article.
-
Culicoides Biting Midges-Underestimated Vectors for Arboviruses of Public Health and Veterinary Importance.Viruses. 2019 Apr 24;11(4):376. doi: 10.3390/v11040376. Viruses. 2019. PMID: 31022868 Free PMC article. Review.
-
Culicoides biting midges: their role as arbovirus vectors.Annu Rev Entomol. 2000;45:307-40. doi: 10.1146/annurev.ento.45.1.307. Annu Rev Entomol. 2000. PMID: 10761580 Review.
Cited by
-
The Role of Hematophagous Arthropods, Other than Mosquitoes and Ticks, in Arbovirus Transmission.Viruses. 2025 Jun 30;17(7):932. doi: 10.3390/v17070932. Viruses. 2025. PMID: 40733550 Free PMC article. Review.
References
-
- Silva A.D.S.D., Delatorre E.O., Leon L.A.A., Azevedo S.S.D.D., Leite T.C.N.F., Paula V.S.D. Propriedades Gerais Dos Vírus. In: Lemos E.R.S.D., Villar L.M., Leon L.A.A., Guimarães M.L., Teixeira S.L.M., Paula V.S.D., editors. Tópicos em Virologia. Editora Fiocruz; Rio de Janeiro, Brazil: 2023. pp. 29–45.
-
- Silva A.D.S.D., Lemos A.S., Alves A.D.R., Marques B.C.L., Ribeiro C.R.D.A., Passaes C.P.B., Santos D.R.L.D., Caetano D.G., Costa E.V.D., Bottino F.D.O., et al. Diagnóstico de Infecções Virais. In: Lemos E.R.S.D., Villar L.M., Leon L.A.A., Guimarães M.L., Teixeira S.L.M., Paula V.S.D., editors. Tópicos Em Virologia. Editora Fiocruz; Rio de Janeiro, Brazil: 2023. pp. 47–86.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
