

The 'Oma's of the Gammas-Cancerogenesis by γ-Herpesviruses
- PMID: 39772235
- PMCID: PMC11680331
- DOI: 10.3390/v16121928
The 'Oma's of the Gammas-Cancerogenesis by γ-Herpesviruses
Abstract
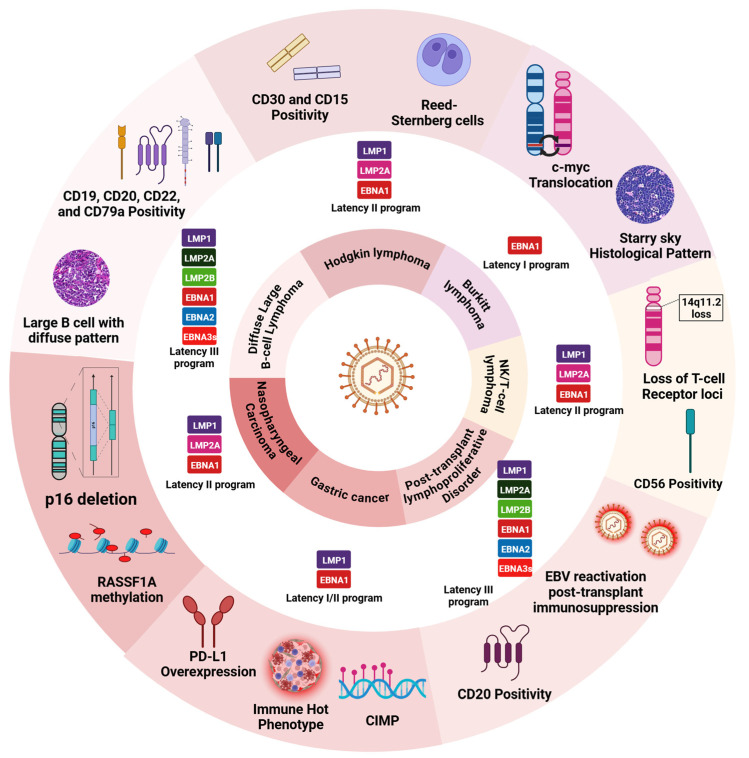
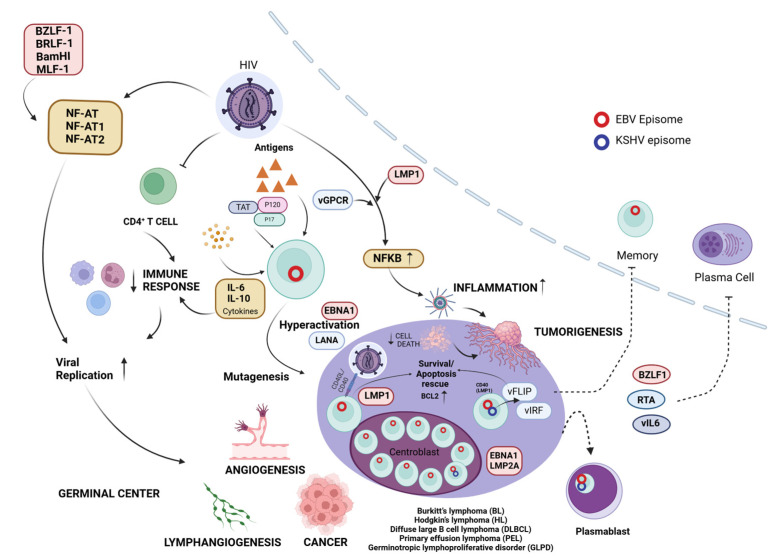
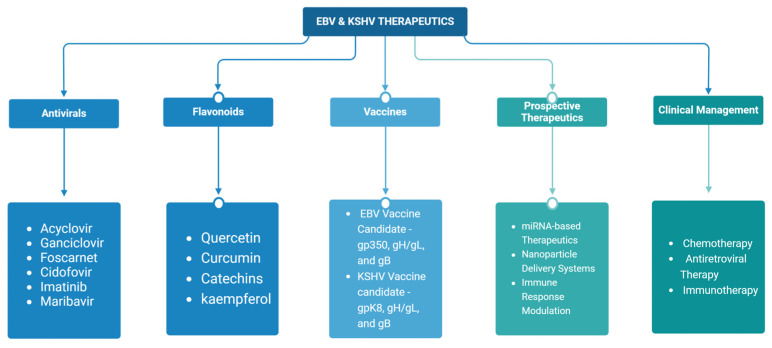
Epstein-Barr virus (EBV) and Kaposi's sarcoma-associated herpesvirus (KSHV), which are the only members of the gamma(γ) herpesviruses, are oncogenic viruses that significantly contribute to the development of various human cancers, such as Burkitt's lymphoma, nasopharyngeal carcinoma, Hodgkin's lymphoma, Kaposi's sarcoma, and primary effusion lymphoma. Oncogenesis triggered by γ-herpesviruses involves complex interactions between viral genetics, host cellular mechanisms, and immune evasion strategies. At the genetic level, crucial viral oncogenes participate in the disruption of cell signaling, leading to uncontrolled proliferation and inhibition of apoptosis. These viral proteins can modulate several cellular pathways, including the NF-κB and JAK/STAT pathways, which play essential roles in cell survival and inflammation. Epigenetic modifications further contribute to EBV- and KSHV-mediated cancerogenesis. Both EBV and KSHV manipulate host cell DNA methylation, histone modification, and chromatin remodeling, the interplay of which contribute to the elevation of oncogene expression and the silencing of the tumor suppressor genes. Immune factors also play a pivotal role in the development of cancer. The γ-herpesviruses have evolved intricate immune evasion strategies, including the manipulation of the major histocompatibility complex (MHC) and the release of cytokines, allowing infected cells to evade immune detection and destruction. In addition, a compromised immune system, such as in HIV/AIDS patients, significantly increases the risk of cancers associated with EBV and KSHV. This review aims to provide a comprehensive overview of the genetic, epigenetic, and immune mechanisms by which γ-herpesviruses drive cancerogenesis, highlighting key molecular pathways and potential therapeutic targets.
Keywords: Epstein–Barr; Kaposi’s sarcoma; antiviral; co-infection; epigenetic; gamma herpesviruses; immune evasion; lymphoma; oncogene; vaccine.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
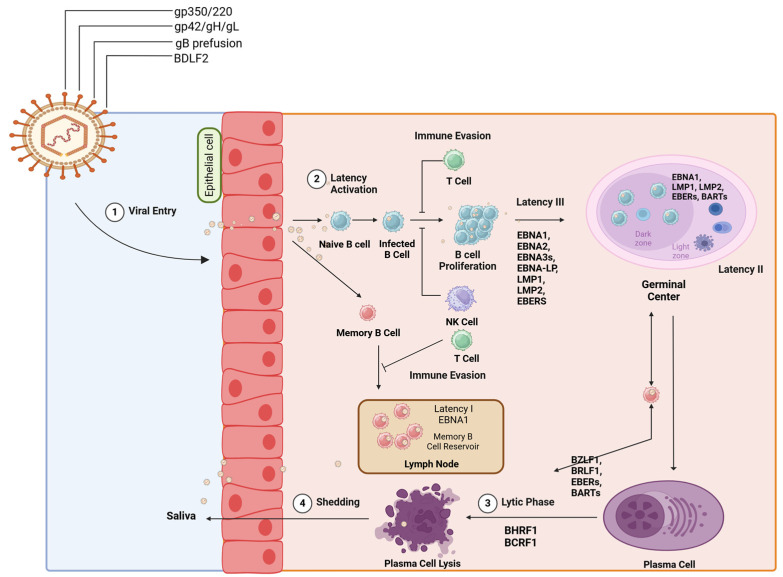
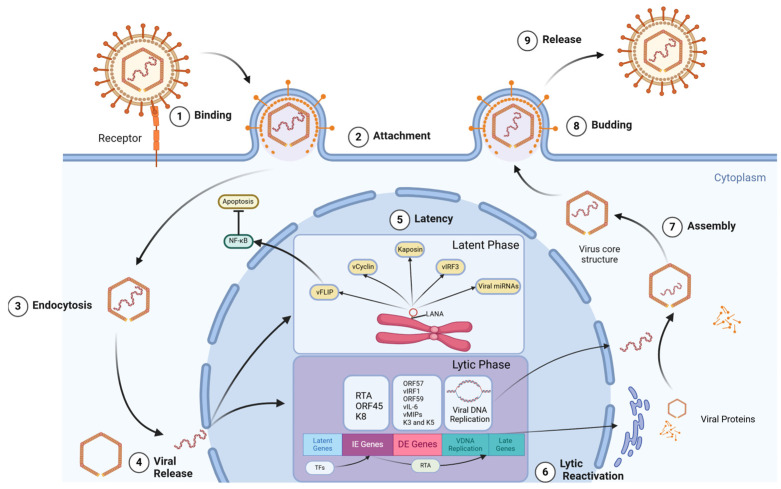
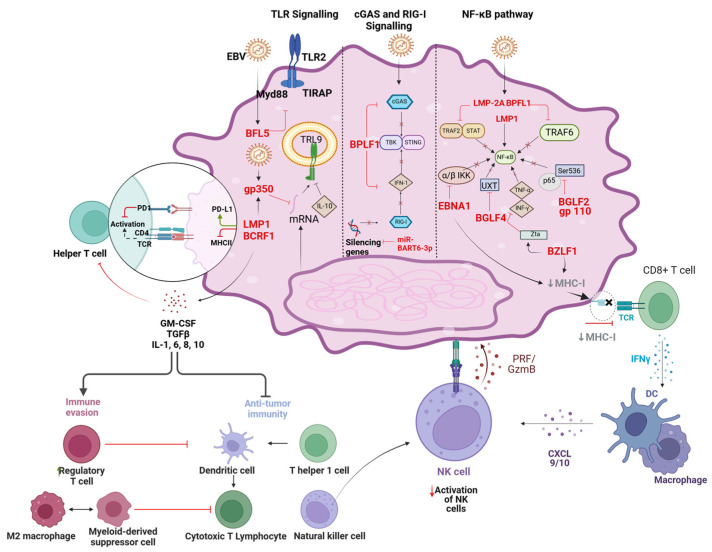
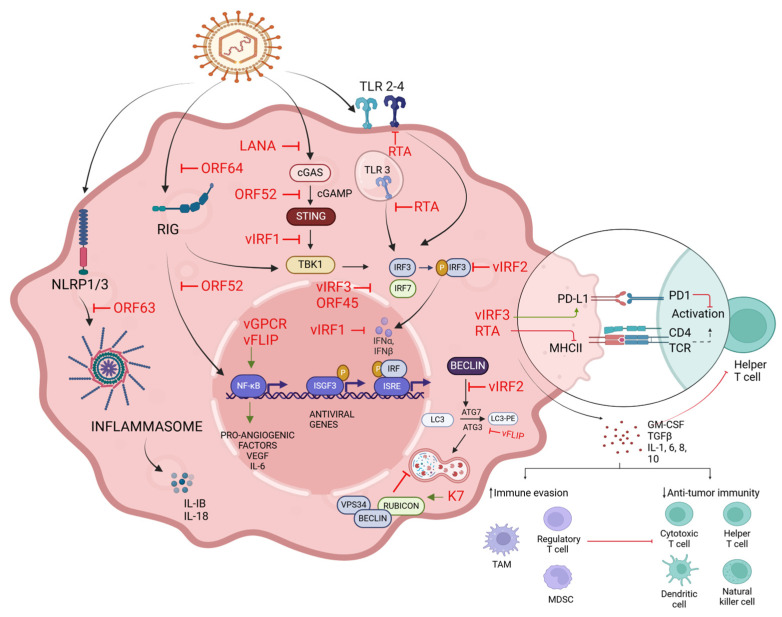
References
-
- Britt W. Virus entry into host, establishment of infection, spread in host, mechanisms of tissue damage. In: Arvin A., Campadelli-Fiume G., Mocarski E., Moore P.S., Roizman B., Whitley R., Yamanishi K., editors. Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis. Cambridge University Press; Cambridge, UK: 2007. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials
