

Targeted sequencing of Enterobacterales bacteria using CRISPR-Cas9 enrichment and Oxford Nanopore Technologies
- PMID: 39772804
- PMCID: PMC11834407
- DOI: 10.1128/msystems.01413-24
Targeted sequencing of Enterobacterales bacteria using CRISPR-Cas9 enrichment and Oxford Nanopore Technologies
Abstract
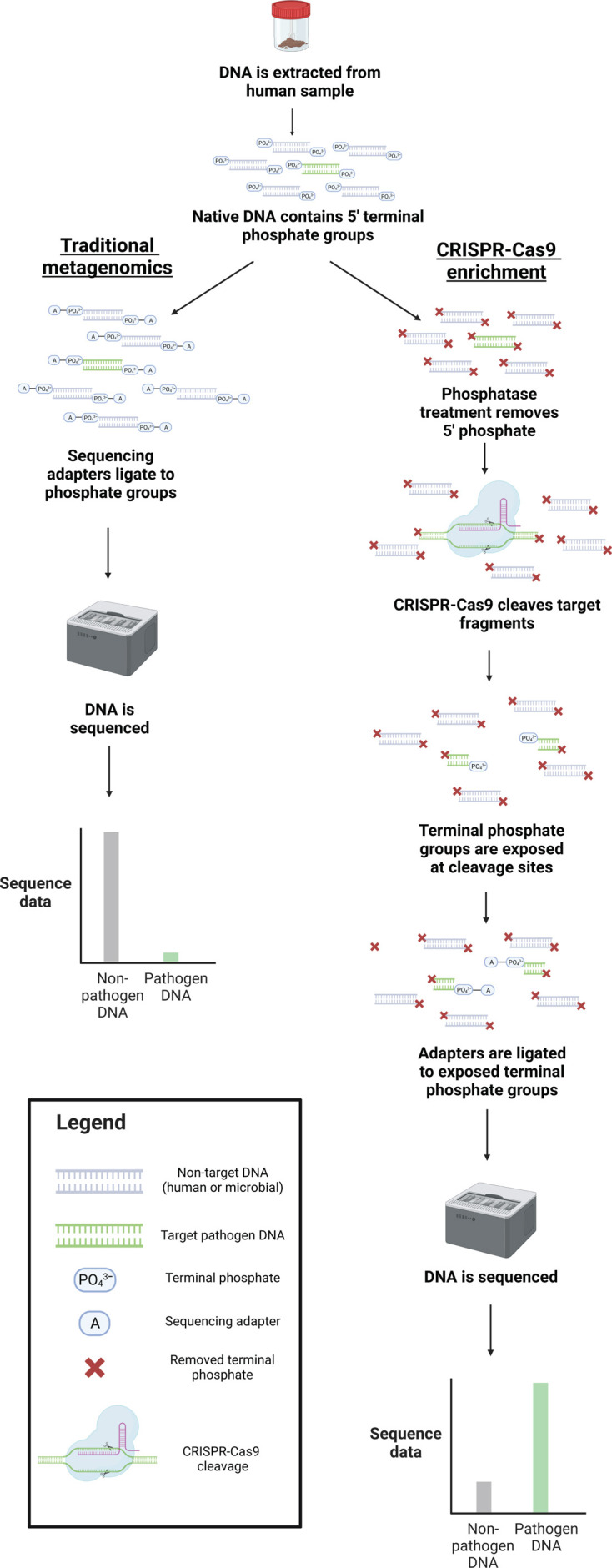
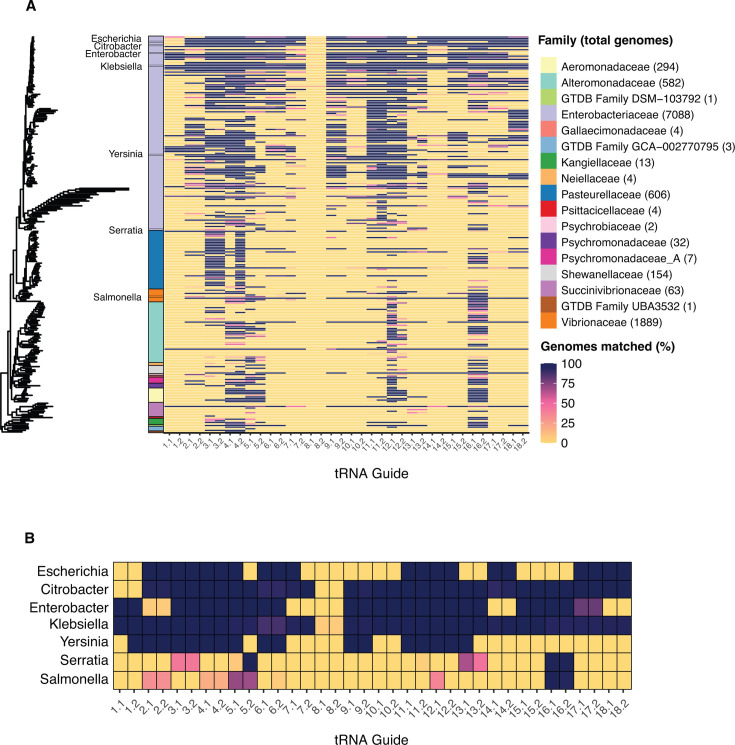
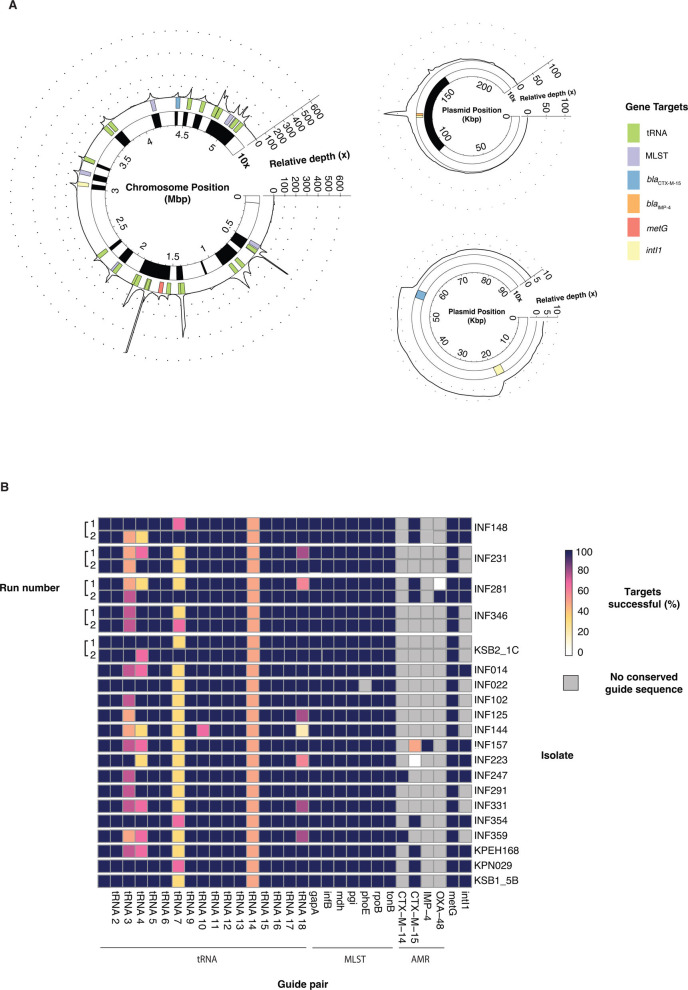
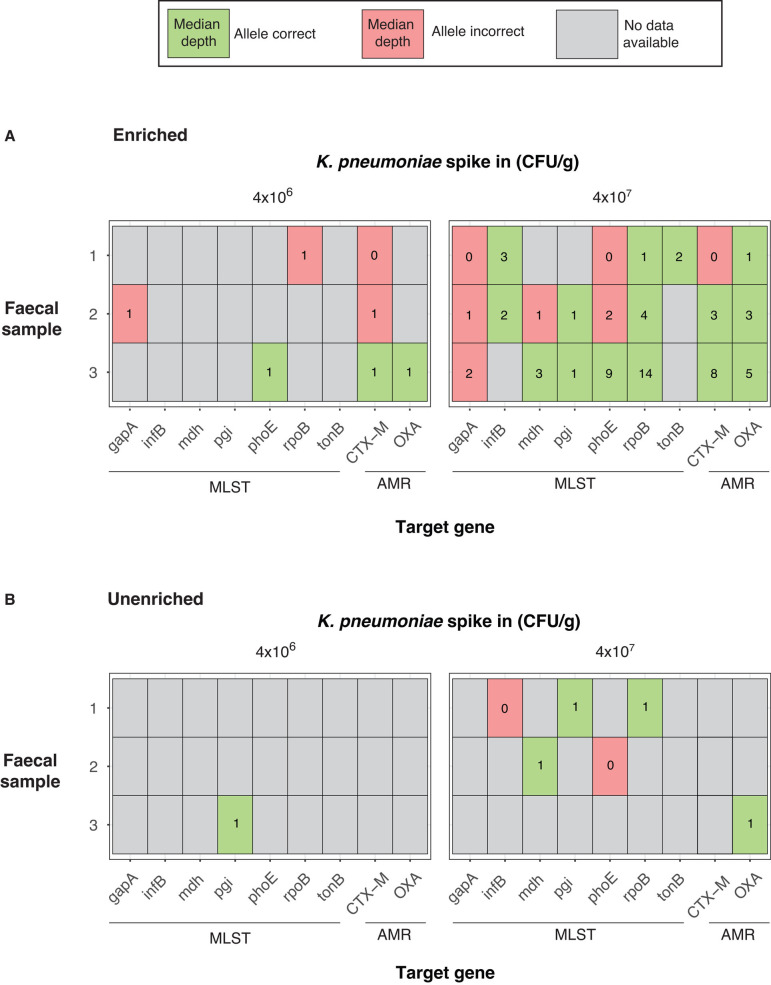
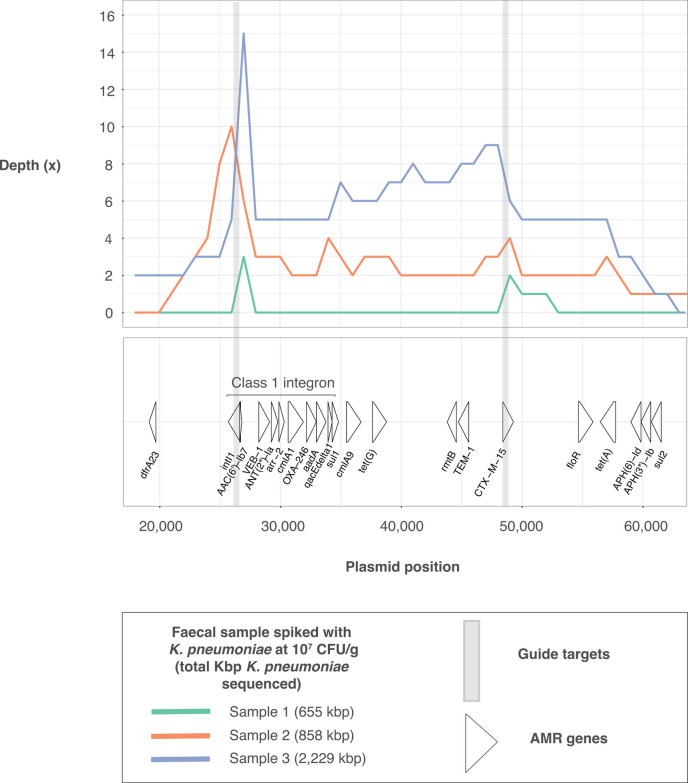
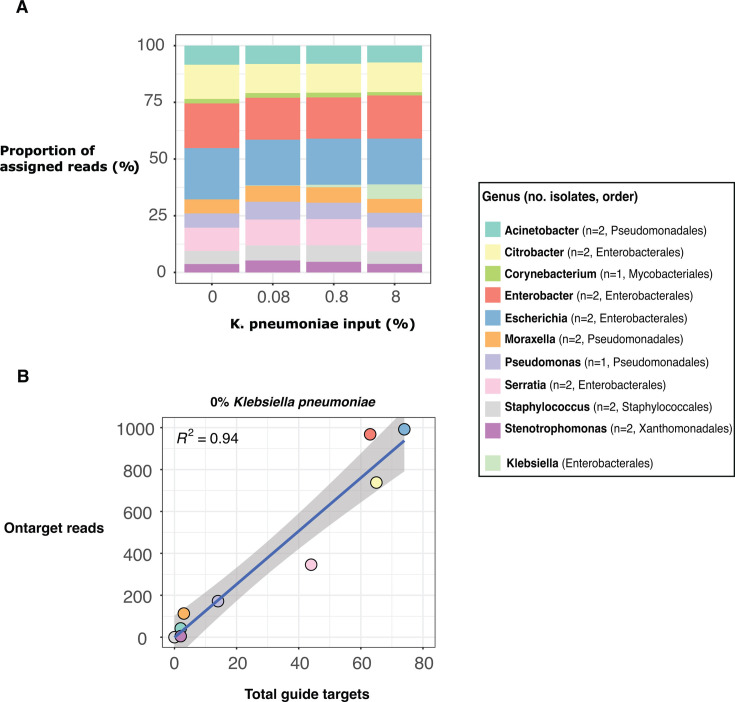
Sequencing DNA directly from patient samples enables faster pathogen characterization compared to traditional culture-based approaches, but often yields insufficient sequence data for effective downstream analysis. CRISPR-Cas9 enrichment is designed to improve the yield of low abundance sequences but has not been thoroughly explored with Oxford Nanopore Technologies (ONT) for use in clinical bacterial epidemiology. We designed CRISPR-Cas9 guide RNAs to enrich the human pathogen Klebsiella pneumoniae, by targeting multi-locus sequence type (MLST) and transfer RNA (tRNA) genes, as well as common antimicrobial resistance (AMR) genes and the resistance-associated integron gene intI1. We validated enrichment performance in 20 K. pneumoniae isolates, finding that guides generated successful enrichment across all conserved sites except for one AMR gene in two isolates. Enrichment of MLST genes led to a correct allele call in all seven loci for 8 out of 10 isolates that had depth of 30× or more in these regions. We then compared enriched and unenriched sequencing of three human fecal samples spiked with K. pneumoniae at varying abundance. Enriched sequencing generated 56× and 11.3× the number of AMR and MLST reads, respectively, compared to unenriched sequencing, and required approximately one-third of the computational storage space. Targeting the intI1 gene often led to detection of 10-20 proximal resistance genes due to the long reads produced by ONT sequencing. We demonstrated that CRISPR-Cas9 enrichment combined with ONT sequencing enabled improved genomic characterization outcomes over unenriched sequencing of patient samples. This method could be used to inform infection control strategies by identifying patients colonized with high-risk strains.
Importance: Understanding bacteria in complex samples can be challenging due to their low abundance, which often results in insufficient data for analysis. To improve the detection of harmful bacteria, we implemented a technique aimed at increasing the amount of data from target pathogens when combined with modern DNA sequencing technologies. Our technique uses CRISPR-Cas9 to target specific gene sequences in the bacterial pathogen Klebsiella pneumoniae and improve recovery from human stool samples. We found our enrichment method to significantly outperform traditional methods, generating far more data originating from our target genes. Additionally, we developed new computational techniques to further enhance the analysis, providing a thorough method for characterizing pathogens from complex biological samples.
Keywords: CRISPR-Cas9 enrichment; Enterobacterales; Klebsiella; Oxford Nanopore; metagenomics.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
