

Multidimensional analyses identify genes of high priority for pancreatic cancer research
- PMID: 39774001
- PMCID: PMC11949049
- DOI: 10.1172/jci.insight.174264
Multidimensional analyses identify genes of high priority for pancreatic cancer research
Abstract
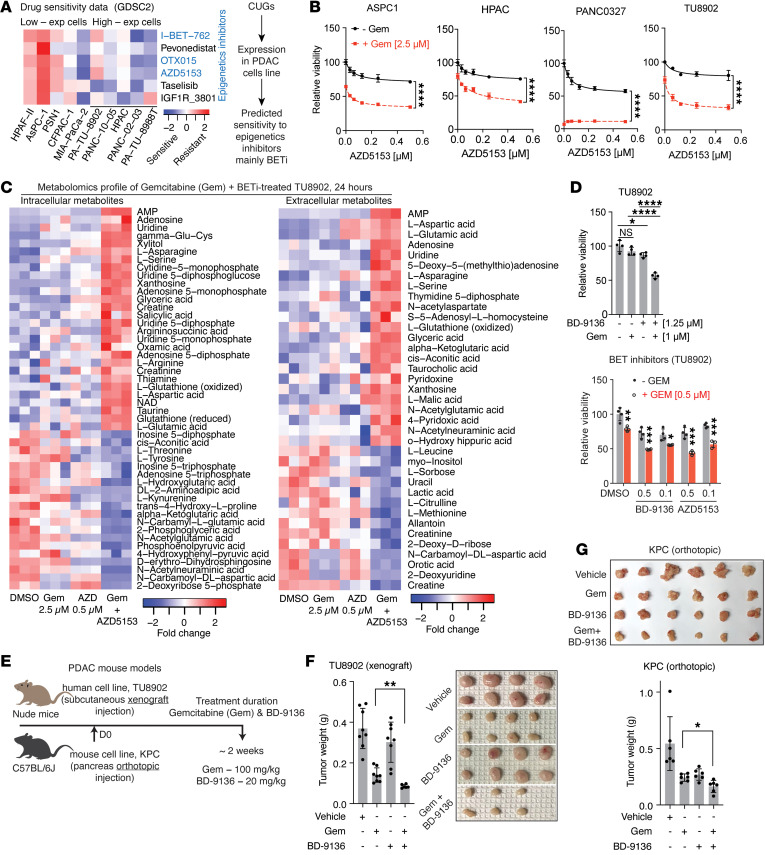
Pancreatic ductal adenocarcinoma (PDAC) is a drug-resistant and lethal cancer. Identification of the genes that consistently show altered expression across patient cohorts can expose effective therapeutic targets and strategies. To identify such genes, we separately analyzed 5 human PDAC microarray datasets. We defined genes as "consistent" if upregulated or downregulated in 4 or more datasets (adjusted P < 0.05). The genes were subsequently queried in additional datasets, including single-cell RNA-sequencing data, and we analyzed their pathway enrichment, tissue specificity, essentiality for cell viability, and association with cancer features, e.g., tumor subtype, proliferation, metastasis, and poor survival outcome. We identified 2,010 consistently upregulated and 1,928 downregulated genes, of which more than 50% to our knowledge were uncharacterized in PDAC. These genes spanned multiple processes, including cell cycle, immunity, transport, metabolism, signaling, and transcriptional/epigenetic regulation - cell cycle and glycolysis being the most altered. Several upregulated genes correlated with cancer features, and their suppression impaired PDAC cell viability in prior CRISPR/Cas9 and RNA interference screens. Furthermore, the upregulated genes predicted sensitivity to bromodomain and extraterminal (epigenetic) protein inhibition, which, in combination with gemcitabine, disrupted amino acid metabolism and in vivo tumor growth. Our results highlight genes for further studies in the quest for PDAC mechanisms, therapeutic targets, and biomarkers.
Keywords: Cancer; Gastroenterology; Glucose metabolism; Molecular genetics; Oncology.
Conflict of interest statement
Figures






Similar articles
-
Regulation of pH by Carbonic Anhydrase 9 Mediates Survival of Pancreatic Cancer Cells With Activated KRAS in Response to Hypoxia.Gastroenterology. 2019 Sep;157(3):823-837. doi: 10.1053/j.gastro.2019.05.004. Epub 2019 May 9. Gastroenterology. 2019. PMID: 31078621
-
S100A14 promotes progression and gemcitabine resistance in pancreatic cancer.Pancreatology. 2021 Apr;21(3):589-598. doi: 10.1016/j.pan.2021.01.011. Epub 2021 Jan 22. Pancreatology. 2021. PMID: 33579599
-
SCNrank: spectral clustering for network-based ranking to reveal potential drug targets and its application in pancreatic ductal adenocarcinoma.BMC Med Genomics. 2020 Apr 3;13(Suppl 5):50. doi: 10.1186/s12920-020-0681-6. BMC Med Genomics. 2020. PMID: 32241274 Free PMC article.
-
BRD4 promotes pancreatic ductal adenocarcinoma cell proliferation and enhances gemcitabine resistance.Oncol Rep. 2015 Apr;33(4):1699-706. doi: 10.3892/or.2015.3774. Epub 2015 Jan 30. Oncol Rep. 2015. PMID: 25647019
-
Integration of Bioinformatics Resources Reveals the Therapeutic Benefits of Gemcitabine and Cell Cycle Intervention in SMAD4-Deleted Pancreatic Ductal Adenocarcinoma.Genes (Basel). 2019 Sep 28;10(10):766. doi: 10.3390/genes10100766. Genes (Basel). 2019. PMID: 31569425 Free PMC article.
Cited by
-
Updates in the diagnosis and management of ductal adenocarcinoma of the pancreas.World J Clin Oncol. 2025 Jun 24;16(6):105601. doi: 10.5306/wjco.v16.i6.105601. World J Clin Oncol. 2025. PMID: 40585824 Free PMC article. Review.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
