

Deep CRISPR mutagenesis characterizes the functional diversity of TP53 mutations
- PMID: 39774325
- PMCID: PMC11735402
- DOI: 10.1038/s41588-024-02039-4
Deep CRISPR mutagenesis characterizes the functional diversity of TP53 mutations
Abstract
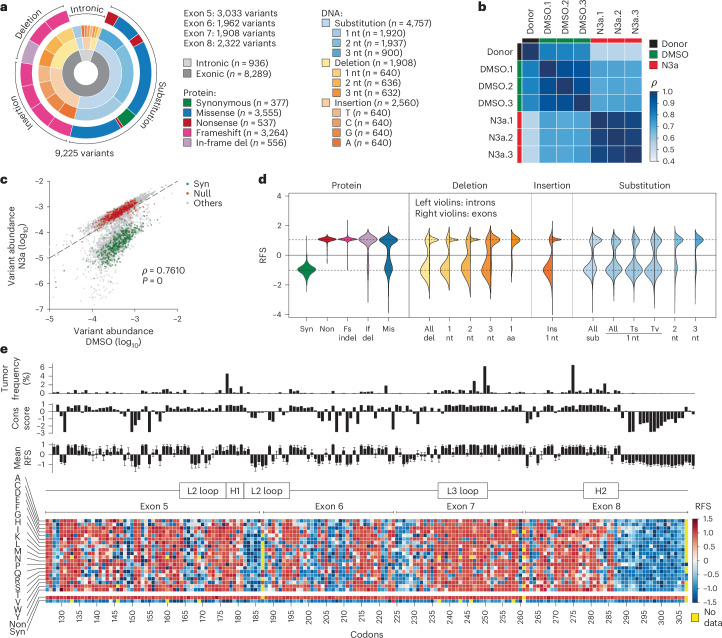
The mutational landscape of TP53, a tumor suppressor mutated in about half of all cancers, includes over 2,000 known missense mutations. To fully leverage TP53 mutation status for personalized medicine, a thorough understanding of the functional diversity of these mutations is essential. We conducted a deep mutational scan using saturation genome editing with CRISPR-mediated homology-directed repair to engineer 9,225 TP53 variants in cancer cells. This high-resolution approach, covering 94.5% of all cancer-associated TP53 missense mutations, precisely mapped the impact of individual mutations on tumor cell fitness, surpassing previous deep mutational scan studies in distinguishing benign from pathogenic variants. Our results revealed even subtle loss-of-function phenotypes and identified promising mutants for pharmacological reactivation. Moreover, we uncovered the roles of splicing alterations and nonsense-mediated messenger RNA decay in mutation-driven TP53 dysfunction. These findings underscore the power of saturation genome editing in advancing clinical TP53 variant interpretation for genetic counseling and personalized cancer therapy.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures

















References
-
- Sabapathy, K. & Lane, D. P. Therapeutic targeting of p53: all mutants are equal, but some mutants are more equal than others. Nat. Rev. Clin. Oncol.15, 13–30 (2018). - PubMed
-
- Stiewe, T. & Haran, T. E. How mutations shape p53 interactions with the genome to promote tumorigenesis and drug resistance. Drug Resist. Updat.38, 27–43 (2018). - PubMed
MeSH terms
Substances
Grants and funding
- STI 182/15-1/Deutsche Forschungsgemeinschaft (German Research Foundation)
- STI 182/13-1/Deutsche Forschungsgemeinschaft (German Research Foundation)
- GRK2573/Deutsche Forschungsgemeinschaft (German Research Foundation)
- 109546710 Project A10/Deutsche Forschungsgemeinschaft (German Research Foundation)
- SA 1923/7-1/Deutsche Forschungsgemeinschaft (German Research Foundation)
- CRC1213 A10N/Deutsche Forschungsgemeinschaft (German Research Foundation)
- JO 1473/1-3/Deutsche Forschungsgemeinschaft (German Research Foundation)
- 031L0063/Bundesministerium für Bildung und Forschung (Federal Ministry of Education and Research)
- DZL/Bundesministerium für Bildung und Forschung (Federal Ministry of Education and Research)
- LOEWE iCANx/Hessisches Ministerium für Wissenschaft und Kunst (Hessen State Ministry of Higher Education, Research and the Arts)
- 65-0004/Von-Behring-Röntgen-Stiftung (Von Behring-Röntgen-Foundation)
- 66-LV06/Von-Behring-Röntgen-Stiftung (Von Behring-Röntgen-Foundation)
- 71-0012/Von-Behring-Röntgen-Stiftung (Von Behring-Röntgen-Foundation)
- 09 R/2018/José Carreras Leukämie-Stiftung (Deutsche José Carreras Leukämie-Stiftung)
- TACTIC/Deutsche Krebshilfe (German Cancer Aid)
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
