

Prioritization of causal genes from genome-wide association studies by Bayesian data integration across loci
- PMID: 39774334
- PMCID: PMC11741684
- DOI: 10.1371/journal.pcbi.1012725
Prioritization of causal genes from genome-wide association studies by Bayesian data integration across loci
Abstract
Motivation: Genome-wide association studies (GWAS) have identified genetic variants, usually single-nucleotide polymorphisms (SNPs), associated with human traits, including disease and disease risk. These variants (or causal variants in linkage disequilibrium with them) usually affect the regulation or function of a nearby gene. A GWAS locus can span many genes, however, and prioritizing which gene or genes in a locus are most likely to be causal remains a challenge. Better prioritization and prediction of causal genes could reveal disease mechanisms and suggest interventions.
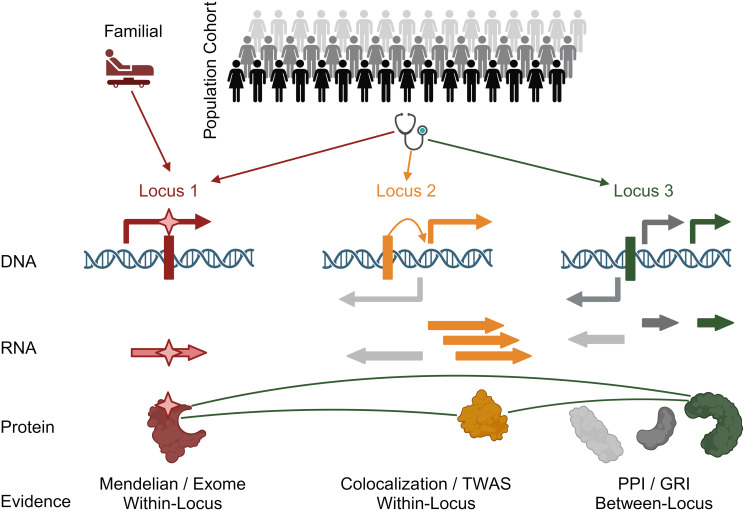
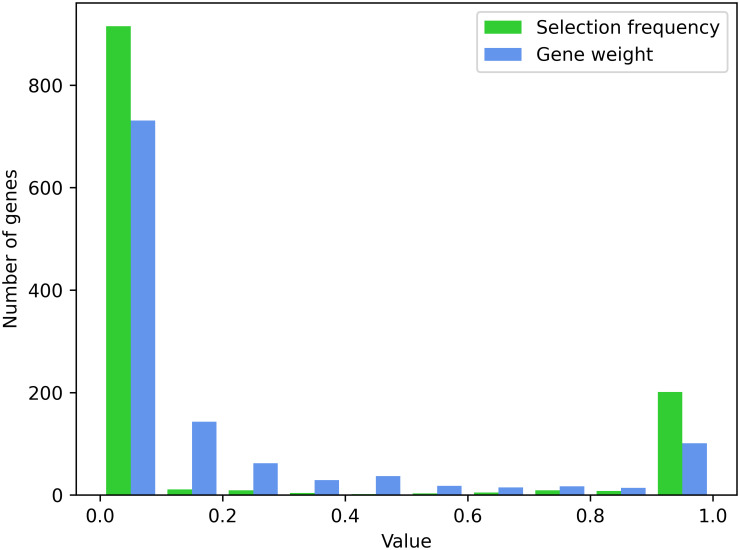
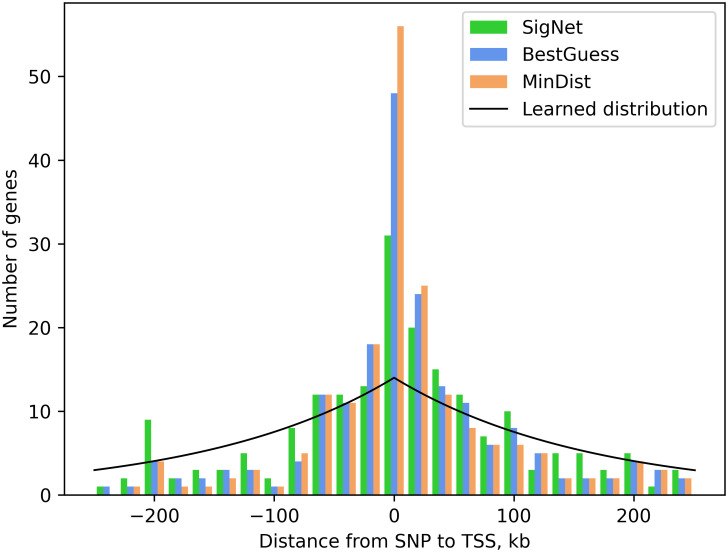
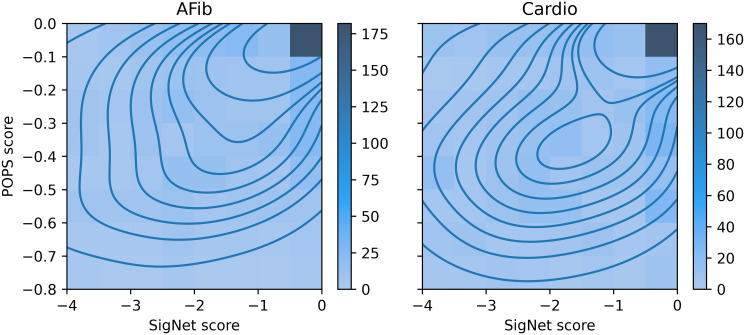
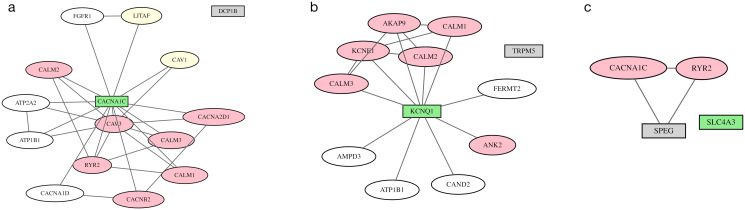
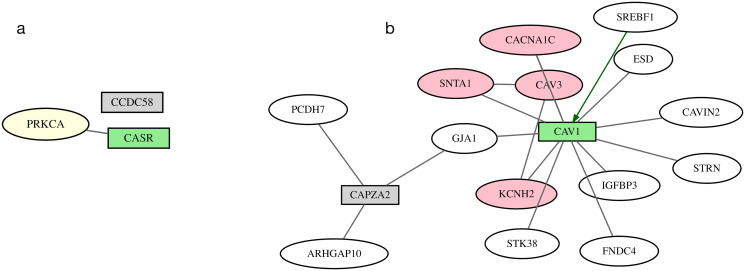
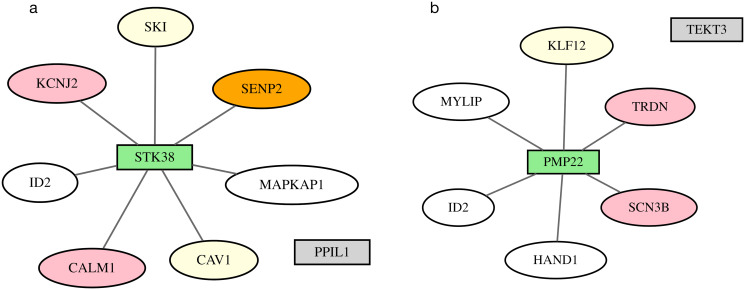
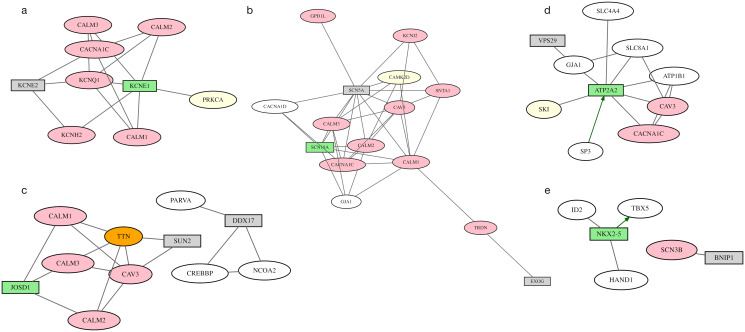
Results: We describe a new Bayesian method, termed SigNet for significance networks, that combines information both within and across loci to identify the most likely causal gene at each locus. The SigNet method builds on existing methods that focus on individual loci with evidence from gene distance and expression quantitative trait loci (eQTL) by sharing information across loci using protein-protein and gene regulatory interaction network data. In an application to cardiac electrophysiology with 226 GWAS loci, only 46 (20%) have within-locus evidence from Mendelian genes, protein-coding changes, or colocalization with eQTL signals. At the remaining 180 loci lacking functional information, SigNet selects 56 genes other than the minimum distance gene, equal to 31% of the information-poor loci and 25% of the GWAS loci overall. Assessment by pathway enrichment demonstrates improved performance by SigNet. Review of individual loci shows literature evidence for genes selected by SigNet, including PMP22 as a novel causal gene candidate.
Copyright: © 2025 Mousavi et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
◦ I have read the journal’s policy and the authors of this manuscript have the following competing interests: JSB is a founder of and advisor to Neochromosome, Inc., and its parent company Opentrons Labworks, Inc. JSB is an advisor to Dextera Biosciences, Inc, has equity in Opentrons and equity and vested options in Dextera.
Figures
Similar articles
-
Colocalization of GWAS and eQTL signals at loci with multiple signals identifies additional candidate genes for body fat distribution.Hum Mol Genet. 2019 Dec 15;28(24):4161-4172. doi: 10.1093/hmg/ddz263. Hum Mol Genet. 2019. PMID: 31691812 Free PMC article.
-
Prediction of causal genes at GWAS loci with pleiotropic gene regulatory effects using sets of correlated instrumental variables.PLoS Genet. 2024 Nov 11;20(11):e1011473. doi: 10.1371/journal.pgen.1011473. eCollection 2024 Nov. PLoS Genet. 2024. PMID: 39527631 Free PMC article.
-
MRLocus: Identifying causal genes mediating a trait through Bayesian estimation of allelic heterogeneity.PLoS Genet. 2021 Apr 19;17(4):e1009455. doi: 10.1371/journal.pgen.1009455. eCollection 2021 Apr. PLoS Genet. 2021. PMID: 33872308 Free PMC article.
-
From genetic associations to genes: methods, applications, and challenges.Trends Genet. 2024 Aug;40(8):642-667. doi: 10.1016/j.tig.2024.04.008. Epub 2024 May 11. Trends Genet. 2024. PMID: 38734482 Review.
-
Opportunities and challenges for transcriptome-wide association studies.Nat Genet. 2019 Apr;51(4):592-599. doi: 10.1038/s41588-019-0385-z. Epub 2019 Mar 29. Nat Genet. 2019. PMID: 30926968 Free PMC article. Review.
References
-
- McKusick V. Online Mendelian Inheritance in Man, OMIM. McKusick-Nathans Institute for Genetic Medicine, Johns Hopkins University (Baltimore, MD) and National Center for Biotechnology Information, National Library of Medicine (Bethesda, MD), 2000. World Wide Web URL: https://omim.org. 2009;.
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
