

EZH2 modulates mRNA splicing and exerts part of its oncogenic function through repression of splicing factors in CML
- PMID: 39774797
- PMCID: PMC11879851
- DOI: 10.1038/s41375-024-02509-y
EZH2 modulates mRNA splicing and exerts part of its oncogenic function through repression of splicing factors in CML
Abstract
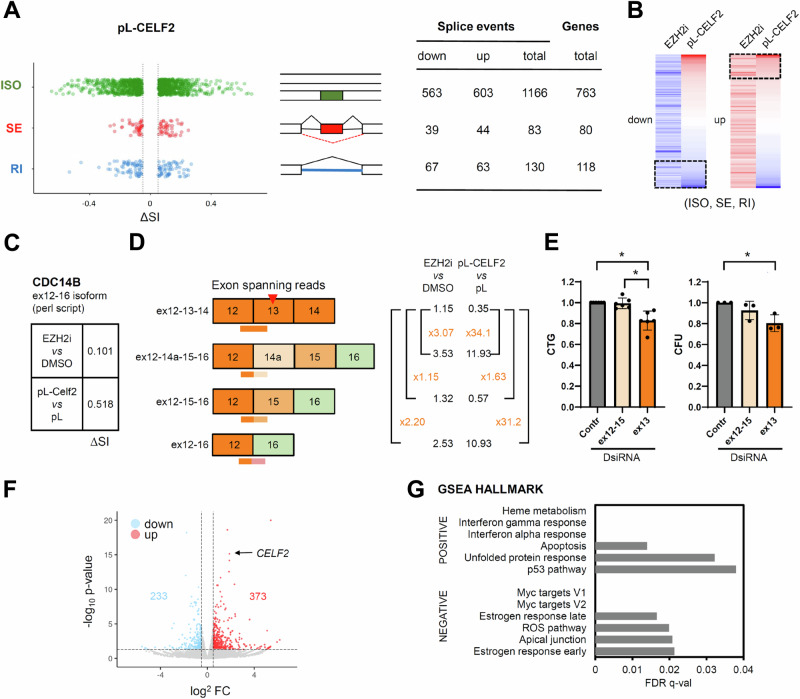
The polycomb protein EZH2 is up-regulated in Chronic Myeloid Leukaemia (CML) and associated with transcriptional reprogramming. Here we tested whether EZH2 might also act as a modulator of the mRNA splicing landscape to elicit its oncogenic function in CML. We treated CML cell lines with EZH2 inhibitors and detected differential splicing of several hundreds of events, potentially caused by the transcriptional regulation of splicing factors. Amongst those genes, CELF2 was identified as a candidate to mediate part of the EZH2 inhibitor induced phenotype. Upon over-expression, we observed (1) reduced cell growth, viability, and colony formation of CML cell lines, (2) a change in the splicing landscape, partially overlapping with EZH2 mediated changes, (3) the down-regulation of MYC signalling. Importantly, these findings were successfully validated in a cohort of CML patient samples, confirming the role of CELF2 as EZH2-regulated tumour-suppressor, contributing to the severe splicing de-regulation present in CML. Based on this we propose that EZH2 exerts part of its oncogenic function in CML through the transcriptional repression of splicing factors. Finally, analysis of publicly available datasets suggests that splicing modulation by EZH2 might not be restricted to CML.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests. Ethics approval and consent to participate: The research was approved by the institutional review board (National University Hospital, DSRB# 2015/00804; DSRB#: 2007/00173) and all human participants gave written informed consent. All methods were performed in accordance with the relevant guidelines and regulations.
Figures






References
-
- Tellier M, Maudlin I, Murphy S. Transcription and splicing: a two-way street. Wiley Interdiscip Rev RNA. 2020;11:e1593. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
