

Improving polygenic prediction from summary data by learning patterns of effect sharing across multiple phenotypes
- PMID: 39775068
- PMCID: PMC11741642
- DOI: 10.1371/journal.pgen.1011519
Improving polygenic prediction from summary data by learning patterns of effect sharing across multiple phenotypes
Abstract
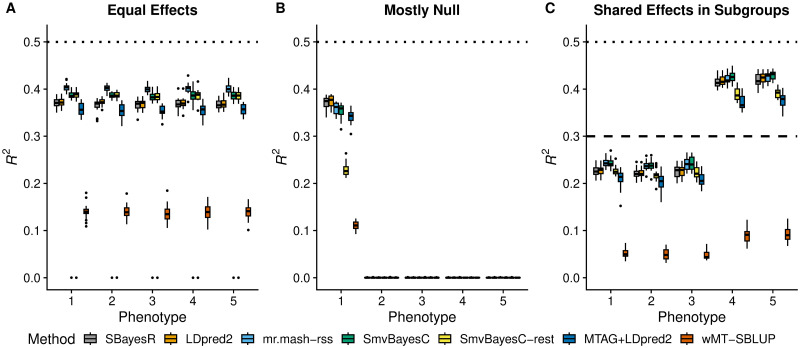
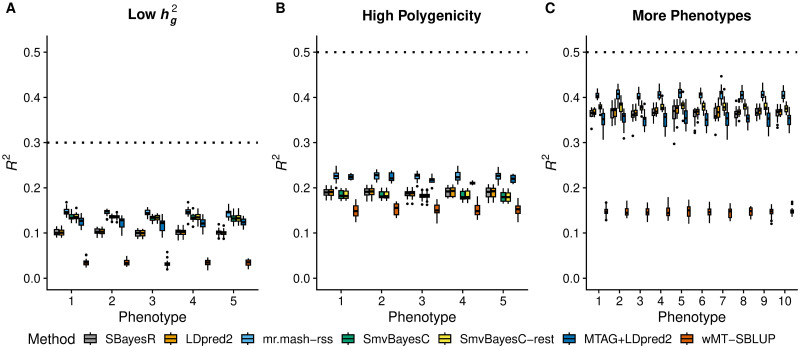
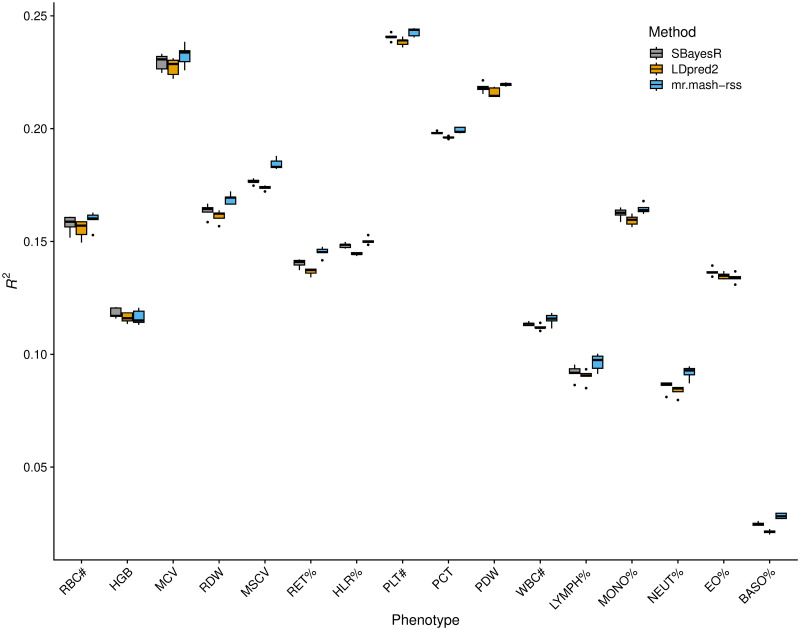
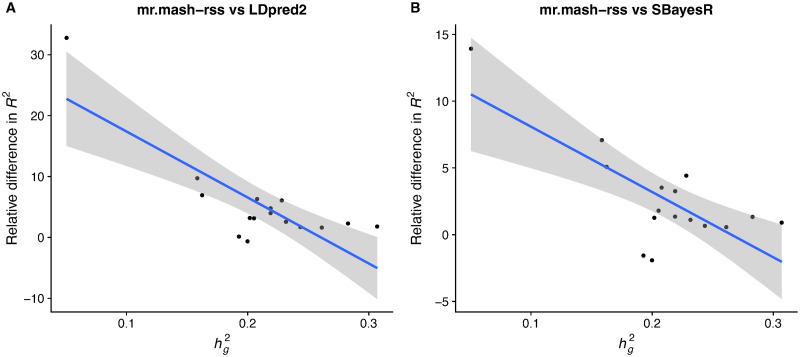
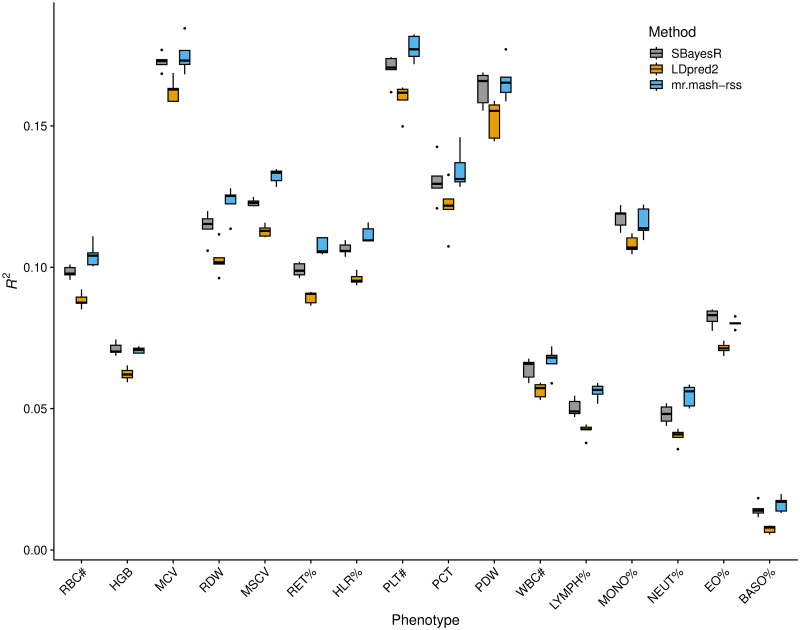
Polygenic prediction of complex trait phenotypes has become important in human genetics, especially in the context of precision medicine. Recently, mr.mash, a flexible and computationally efficient method that models multiple phenotypes jointly and leverages sharing of effects across such phenotypes to improve prediction accuracy, was introduced. However, a drawback of mr.mash is that it requires individual-level data, which are often not publicly available. In this work, we introduce mr.mash-rss, an extension of the mr.mash model that requires only summary statistics from Genome-Wide Association Studies (GWAS) and linkage disequilibrium (LD) estimates from a reference panel. By using summary data, we achieve the twin goal of increasing the applicability of the mr.mash model to data sets that are not publicly available and making it scalable to biobank-size data. Through simulations, we show that mr.mash-rss is competitive with, and often outperforms, current state-of-the-art methods for single- and multi-phenotype polygenic prediction in a variety of scenarios that differ in the pattern of effect sharing across phenotypes, the number of phenotypes, the number of causal variants, and the genomic heritability. We also present a real data analysis of 16 blood cell phenotypes in the UK Biobank, showing that mr.mash-rss achieves higher prediction accuracy than competing methods for the majority of traits, especially when the data set has smaller sample size.
Copyright: © 2025 Kunkel et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Update of
-
Improving polygenic prediction from summary data by learning patterns of effect sharing across multiple phenotypes.bioRxiv [Preprint]. 2024 May 10:2024.05.06.592745. doi: 10.1101/2024.05.06.592745. bioRxiv. 2024. Update in: PLoS Genet. 2025 Jan 07;21(1):e1011519. doi: 10.1371/journal.pgen.1011519. PMID: 38766136 Free PMC article. Updated. Preprint.
References
-
- Walsh B, Lynch M. Evolution and selection of quantitative traits. Oxford University Press; 2018.
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
