

Systematic benchmarking of deep-learning methods for tertiary RNA structure prediction
- PMID: 39775239
- PMCID: PMC11723642
- DOI: 10.1371/journal.pcbi.1012715
Systematic benchmarking of deep-learning methods for tertiary RNA structure prediction
Abstract
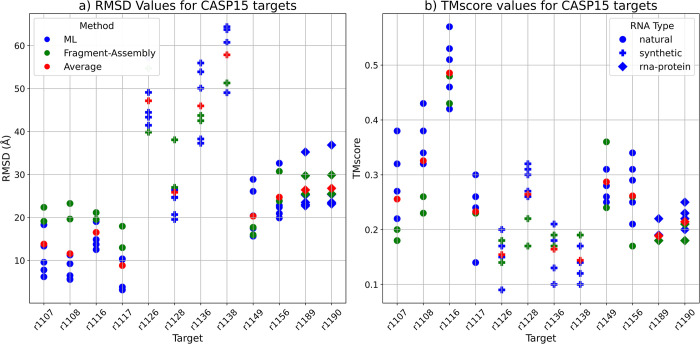
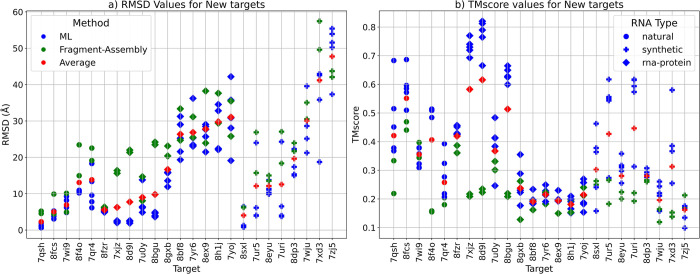
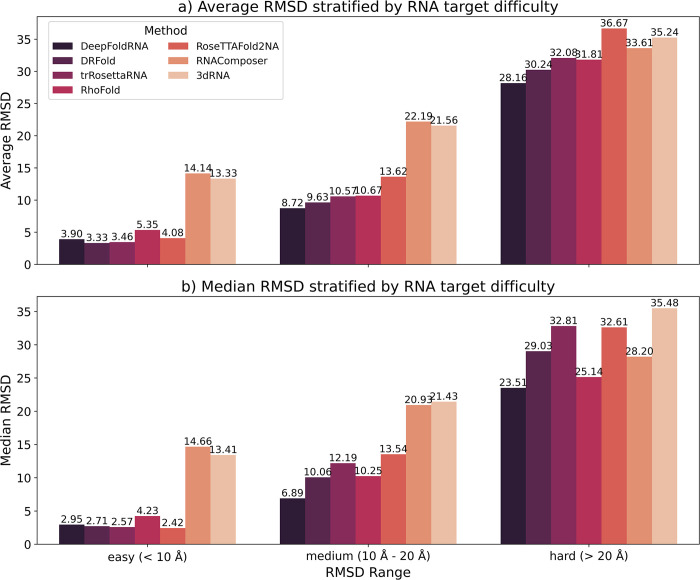
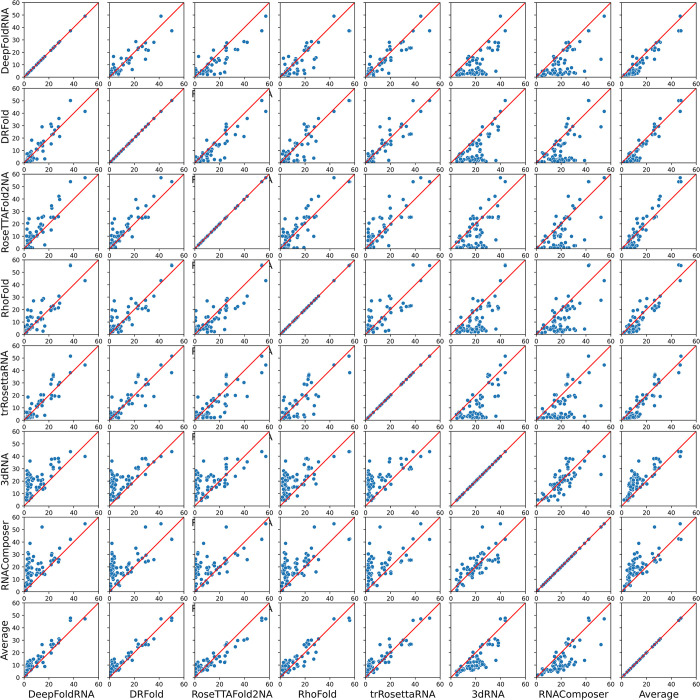
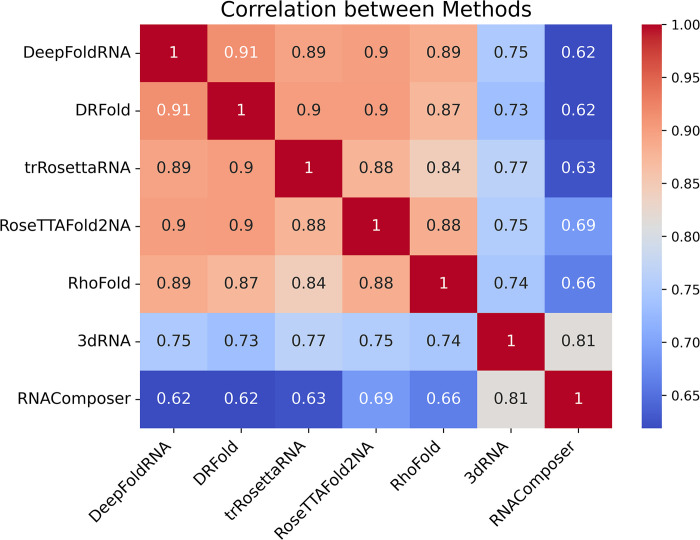
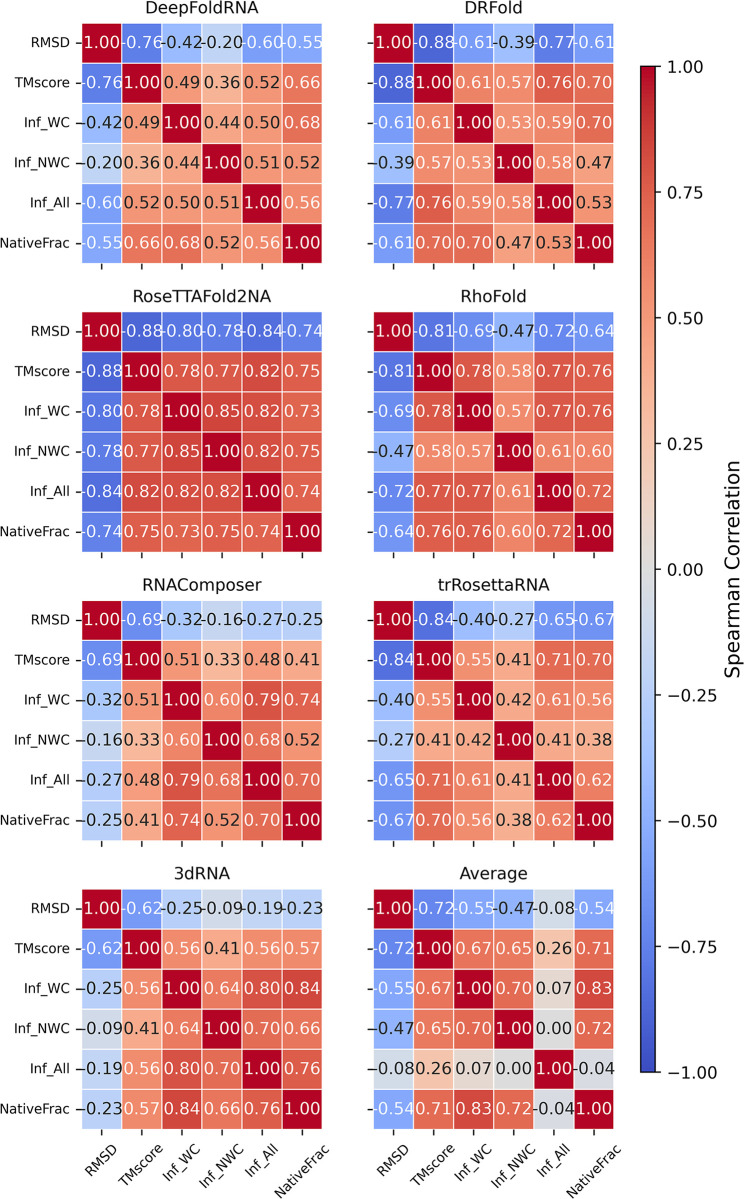
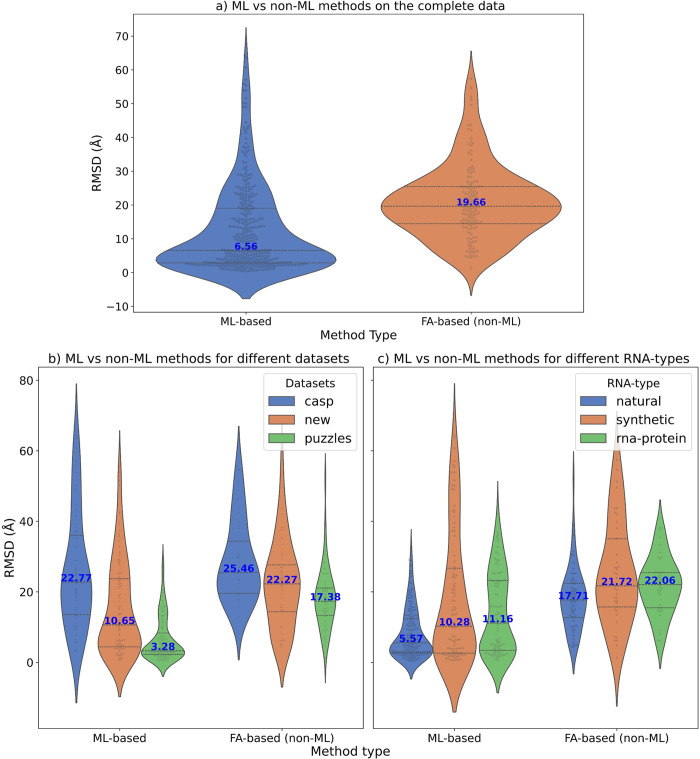
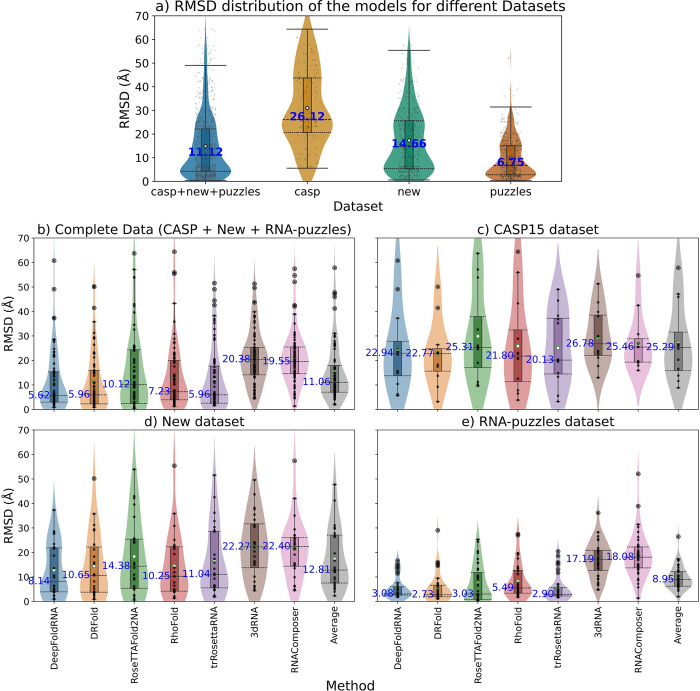
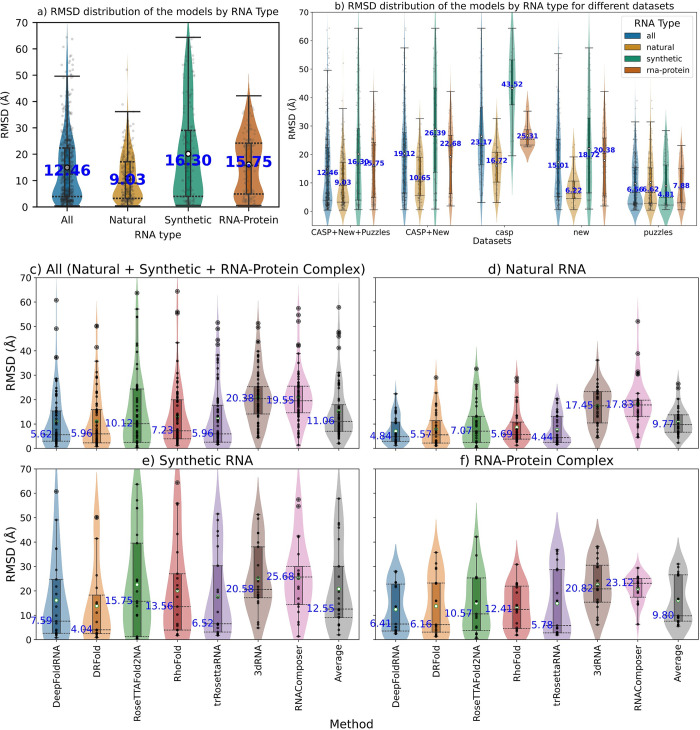
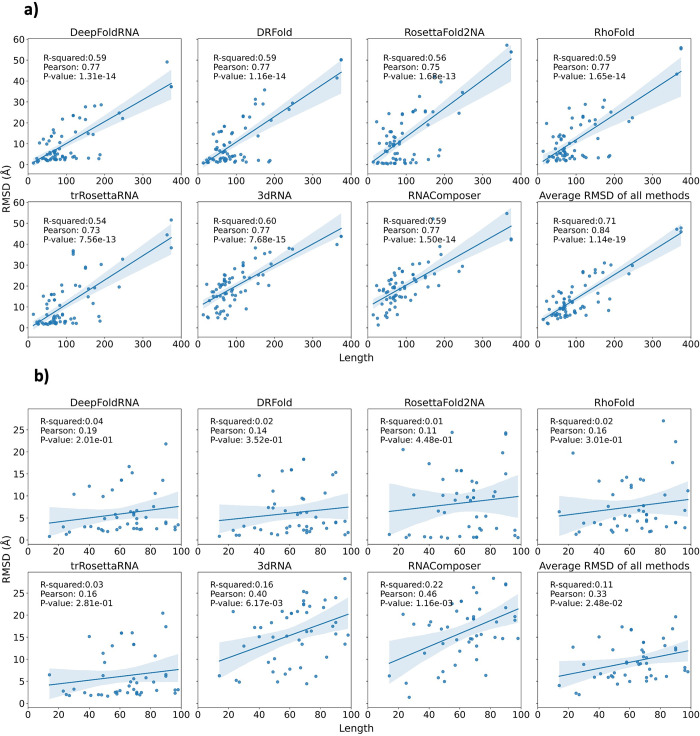
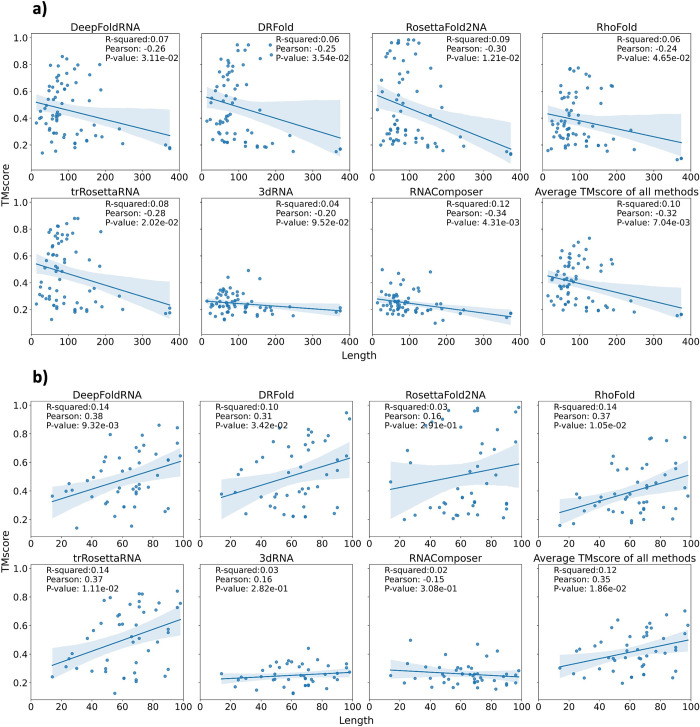
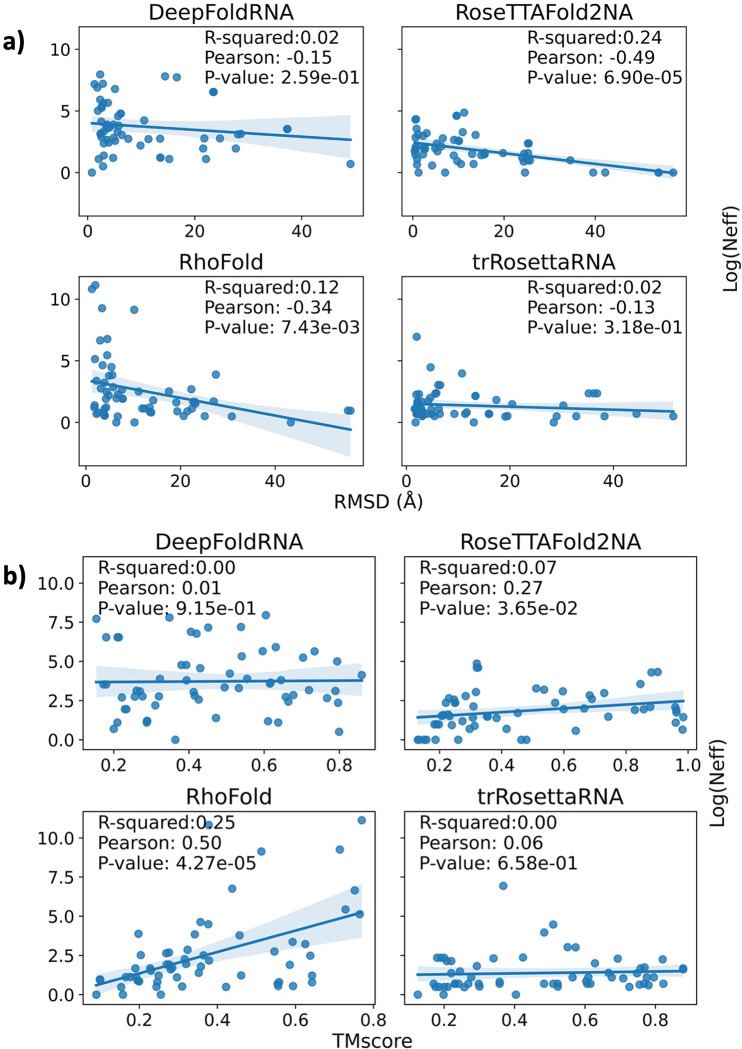
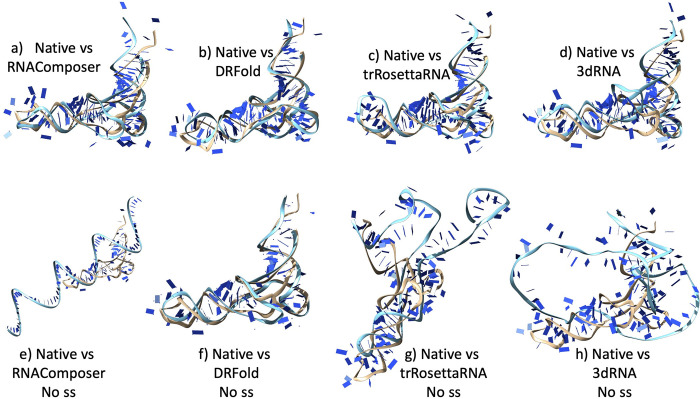
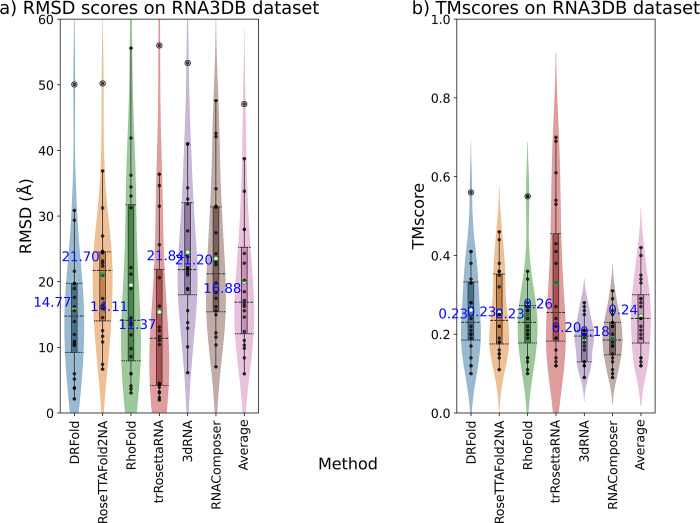
The 3D structure of RNA critically influences its functionality, and understanding this structure is vital for deciphering RNA biology. Experimental methods for determining RNA structures are labour-intensive, expensive, and time-consuming. Computational approaches have emerged as valuable tools, leveraging physics-based-principles and machine learning to predict RNA structures rapidly. Despite advancements, the accuracy of computational methods remains modest, especially when compared to protein structure prediction. Deep learning methods, while successful in protein structure prediction, have shown some promise for RNA structure prediction as well, but face unique challenges. This study systematically benchmarks state-of-the-art deep learning methods for RNA structure prediction across diverse datasets. Our aim is to identify factors influencing performance variation, such as RNA family diversity, sequence length, RNA type, multiple sequence alignment (MSA) quality, and deep learning model architecture. We show that generally ML-based methods perform much better than non-ML methods on most RNA targets, although the performance difference isn't substantial when working with unseen novel or synthetic RNAs. The quality of the MSA and secondary structure prediction both play an important role and most methods aren't able to predict non-Watson-Crick pairs in the RNAs. Overall among the automated 3D RNA structure prediction methods, DeepFoldRNA has the best prediction results followed by DRFold as the second best method. Finally, we also suggest possible mitigations to improve the quality of the prediction for future method development.
Copyright: © 2024 Bahai et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors have declared no competing interests exist.
Figures
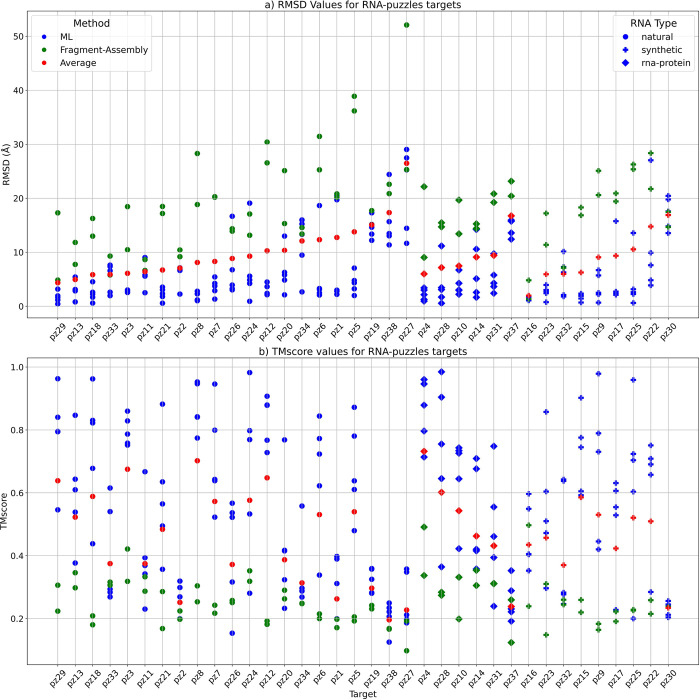
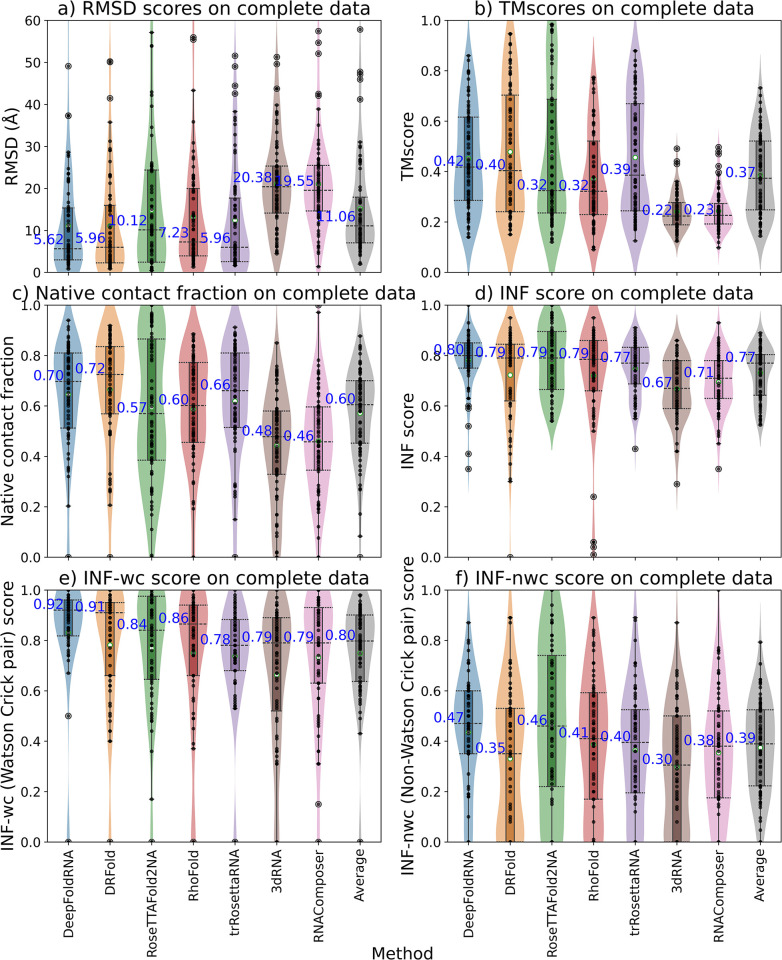
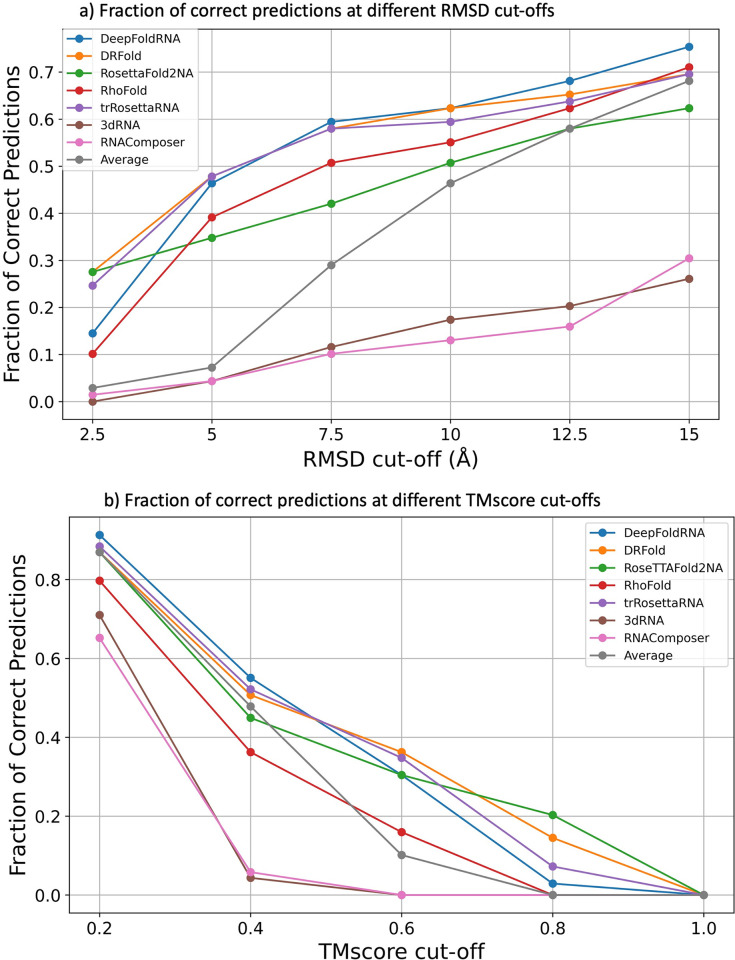
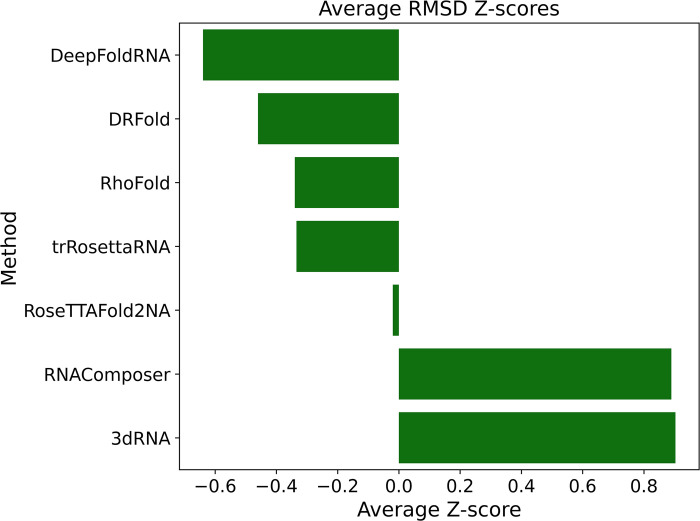
References
-
- Alberts B, Johnson A, Lewis J, Morgan D, Raff MC, Roberts K, et al. Molecular biology of the cell. Sixth edition. New York, NY: Garland Science, Taylor and Francis Group; 2015. 1 p.
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
