

The etiology and prevention of early-stage tau pathology in higher cortical circuits: Insights from aging rhesus macaques
- PMID: 39776253
- PMCID: PMC11848412
- DOI: 10.1002/alz.14477
The etiology and prevention of early-stage tau pathology in higher cortical circuits: Insights from aging rhesus macaques
Abstract
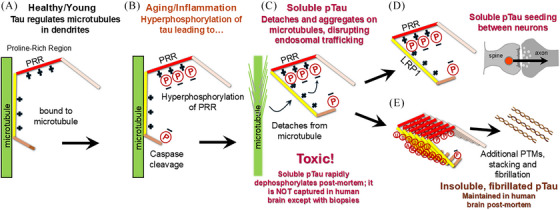
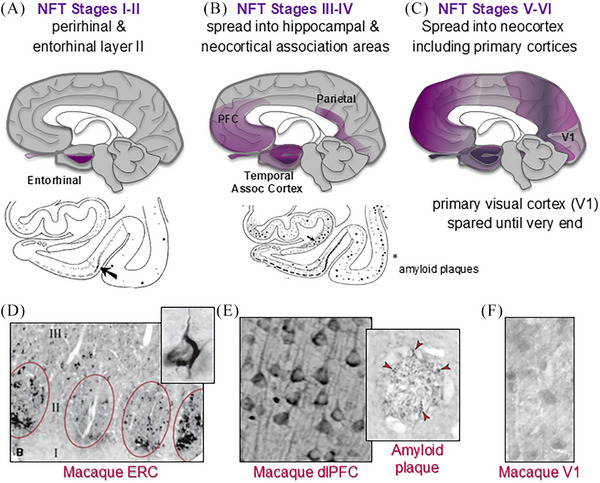
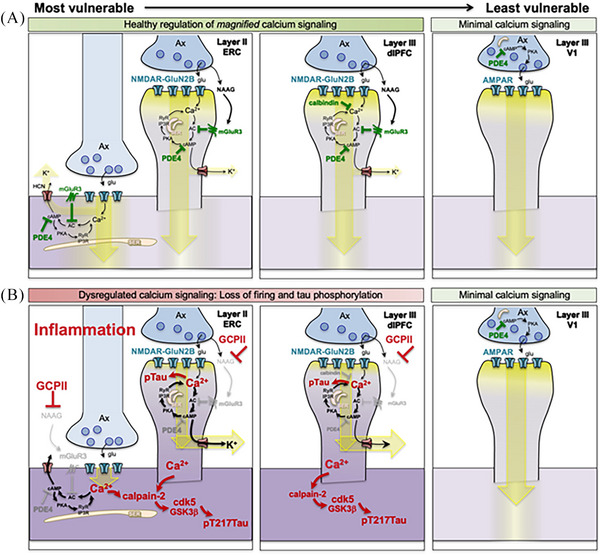
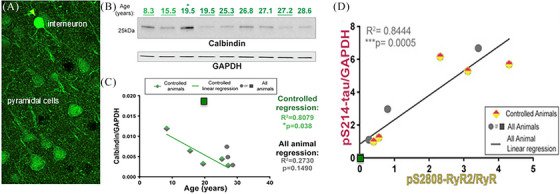
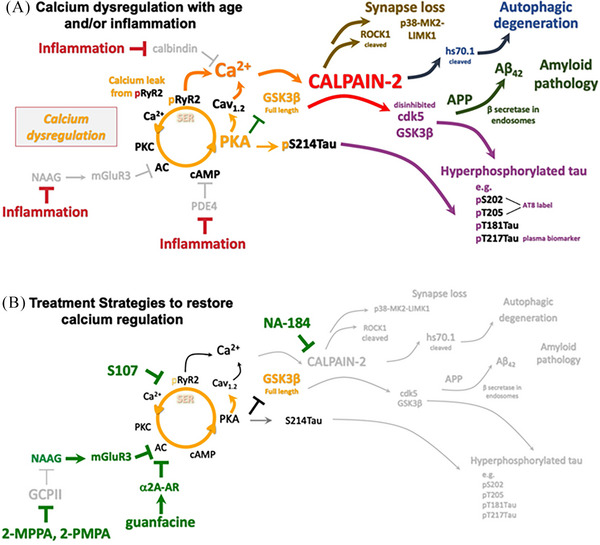
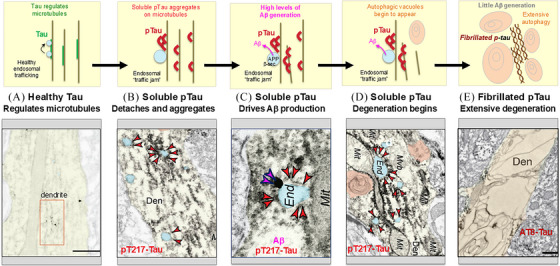
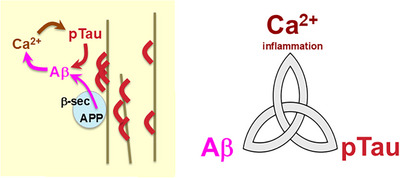
Aging rhesus macaques provide a unique model for learning how age and inflammation drive early-stage pathology in sporadic Alzheimer's disease, and for testing potential therapeutics. Unlike mice, aging macaques have extensive association cortices and inflammatory signaling similar to humans, are apolipoprotein E ε4 homozygotes, and naturally develop tau and amyloid pathology with marked cognitive deficits. Importantly, monkeys provide the unique opportunity to study early-stage, soluble hyperphosphorylated tau (p-tau), including p-tau217. As soluble p-tau is rapidly dephosphorylated post mortem, it is not captured in human brains except with biopsy material. However, new macaque data show that soluble p-tau is toxic to neurons and capable of seeding across cortical circuits. Extensive evidence indicates that age-related inflammatory signaling contributes to calcium dysregulation, which drives tau hyperphosphorylation and amyloid beta generation. Pharmacological studies in aged macaques suggest that inhibiting inflammation and restoring calcium regulation can reduce tau hyperphosphorylation with minimal side effects, appropriate for potential preventive therapeutics. HIGHLIGHTS: Aging monkeys provide a unique window into early stage, soluble phosphorylated tau (p-tau). Inflammation with advancing age leads to calcium dysregulation, p-tau, and amyloid beta (Aβ). Macaque research shows p-tau undergoes transsynaptic seeding early in the cortex. p-tau traps amyloid precursor protein-containing endosomes, which may increase Aβ and drive vicious cycles. Restoring calcium regulation in cortex reduced p-tau217 levels in aged macaques.
Keywords: Alzheimer's disease; association cortex; calcium dysregulation; calpain; glutamate carboxypeptidase II; inflammation; phosphorylated tau; seeding; tau.
© 2024 The Author(s). Alzheimer's & Dementia published by Wiley Periodicals LLC on behalf of Alzheimer's Association.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures

References
-
- King A. The search for better animal models of Alzheimer's disease. Nature. 2018;559:S13‐15. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
