

Machine learning and molecular docking prediction of potential inhibitors against dengue virus
- PMID: 39776767
- PMCID: PMC11703810
- DOI: 10.3389/fchem.2024.1510029
Machine learning and molecular docking prediction of potential inhibitors against dengue virus
Abstract
Introduction: Dengue Fever continues to pose a global threat due to the widespread distribution of its vector mosquitoes, Aedes aegypti and Aedes albopictus. While the WHO-approved vaccine, Dengvaxia, and antiviral treatments like Balapiravir and Celgosivir are available, challenges such as drug resistance, reduced efficacy, and high treatment costs persist. This study aims to identify novel potential inhibitors of the Dengue virus (DENV) using an integrative drug discovery approach encompassing machine learning and molecular docking techniques.
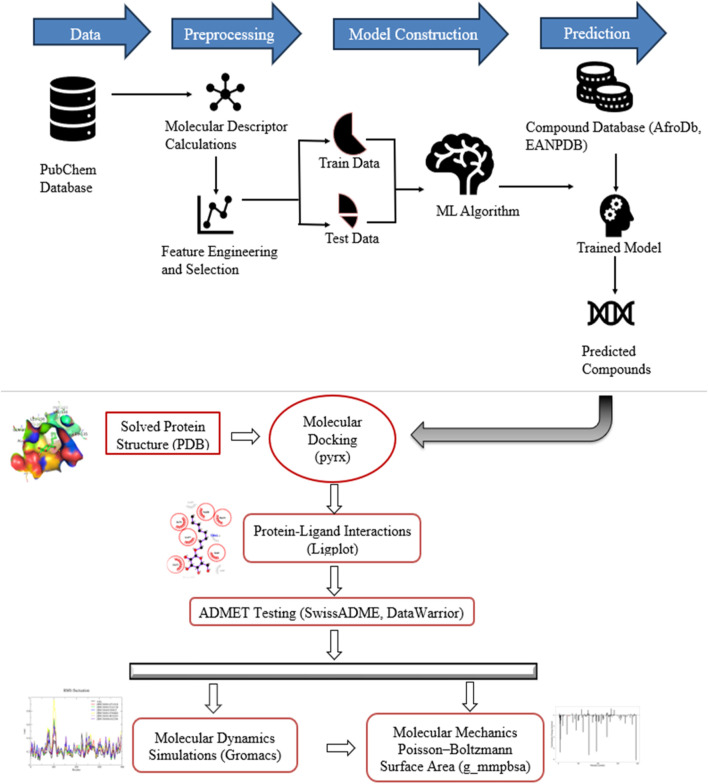
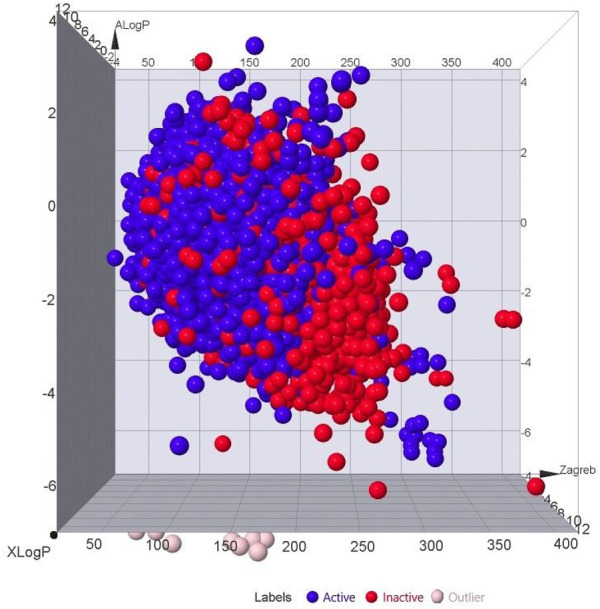
Method: Utilizing a dataset of 21,250 bioactive compounds from PubChem (AID: 651640), alongside a total of 1,444 descriptors generated using PaDEL, we trained various models such as Support Vector Machine, Random Forest, k-nearest neighbors, Logistic Regression, and Gaussian Naïve Bayes. The top-performing model was used to predict active compounds, followed by molecular docking performed using AutoDock Vina. The detailed interactions, toxicity, stability, and conformational changes of selected compounds were assessed through protein-ligand interaction studies, molecular dynamics (MD) simulations, and binding free energy calculations.
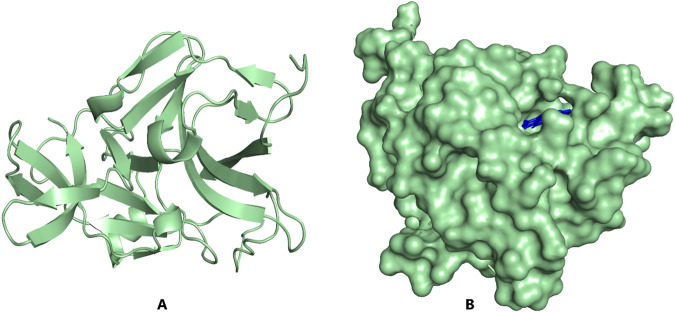
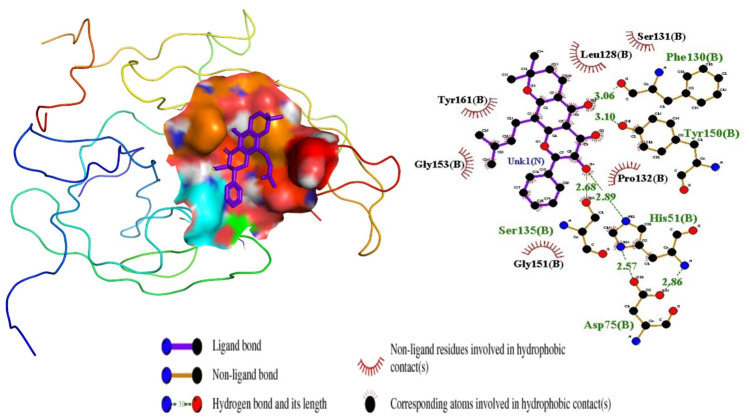
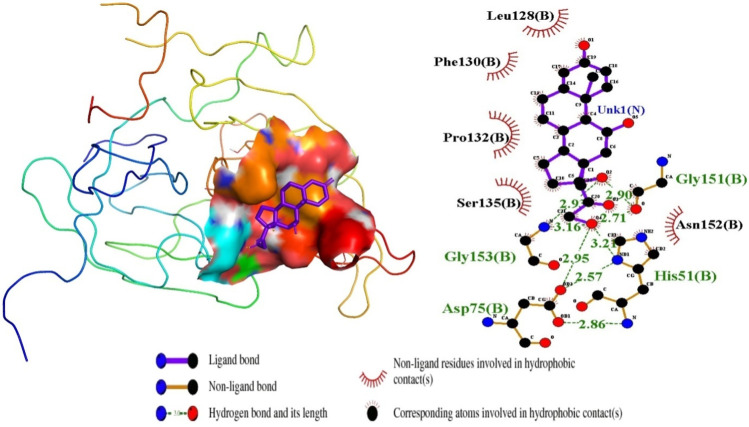
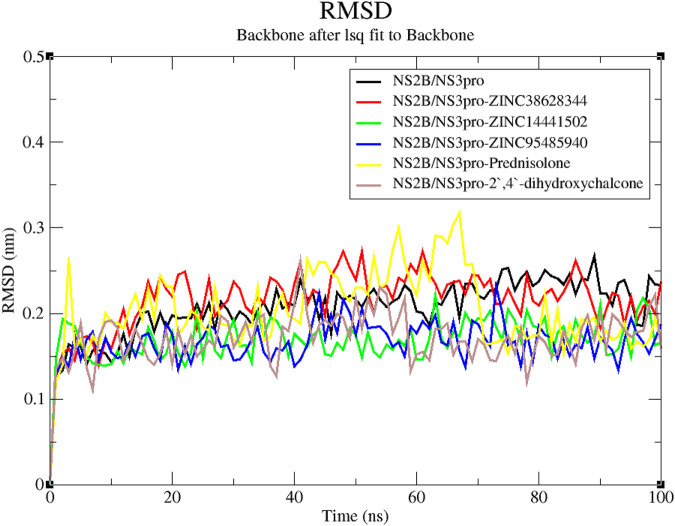
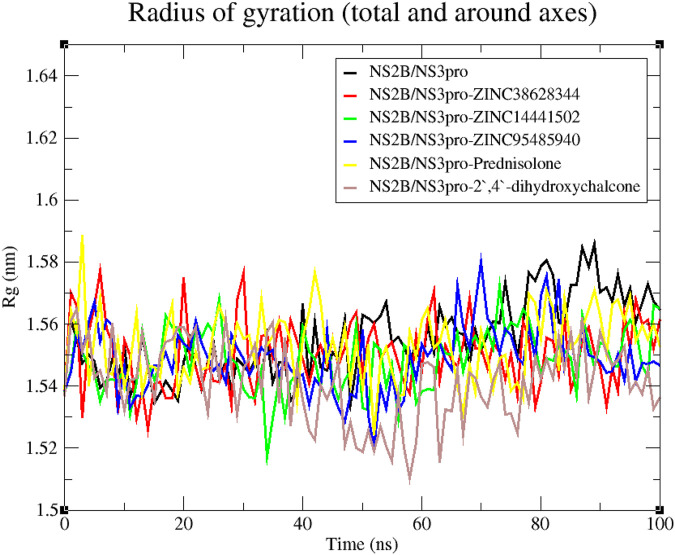
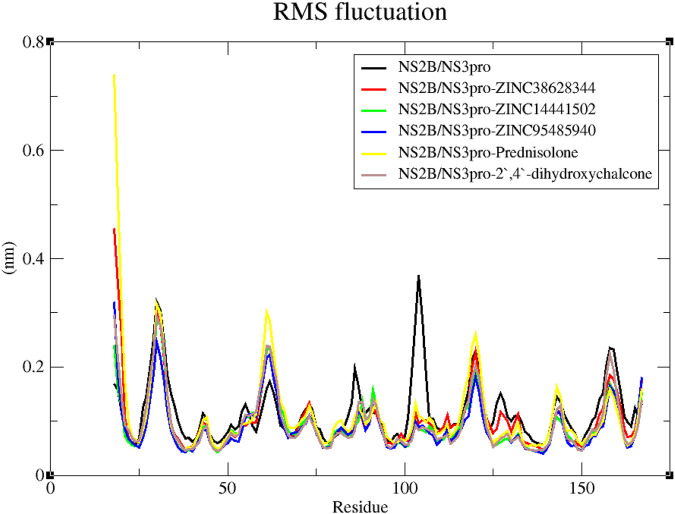
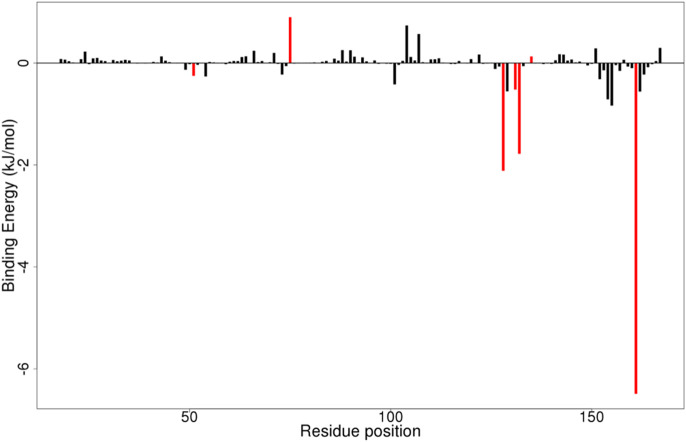
Results: We implemented a robust three-dataset splitting strategy, employing the Logistic Regression algorithm, which achieved an accuracy of 94%. The model successfully predicted 18 known DENV inhibitors, with 11 identified as active, paving the way for further exploration of 2683 new compounds from the ZINC and EANPDB databases. Subsequent molecular docking studies were performed on the NS2B/NS3 protease, an enzyme essential in viral replication. ZINC95485940, ZINC38628344, 2',4'-dihydroxychalcone and ZINC14441502 demonstrated a high binding affinity of -8.1, -8.5, -8.6, and -8.0 kcal/mol, respectively, exhibiting stable interactions with His51, Ser135, Leu128, Pro132, Ser131, Tyr161, and Asp75 within the active site, which are critical residues involved in inhibition. Molecular dynamics simulations coupled with MMPBSA further elucidated the stability, making it a promising candidate for drug development.
Conclusion: Overall, this integrative approach, combining machine learning, molecular docking, and dynamics simulations, highlights the strength and utility of computational tools in drug discovery. It suggests a promising pathway for the rapid identification and development of novel antiviral drugs against DENV. These in silico findings provide a strong foundation for future experimental validations and in-vitro studies aimed at fighting DENV.
Keywords: dengue virus; drug discovery; machine learning; molecular docking; molecular dynamics simulation.
Copyright © 2024 Hanson, Adams, Kepgang, Zondagh, Tem Bueh, Asante, Shirolkar, Kisaakye, Bondarwad and Awe.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Abraham M. J., Murtola T., Schulz R., Pall S., Smith J. C., Hess B., et al. (2015). Gromacs: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25. 10.1016/j.softx.2015.06.001 - DOI
-
- Adams L., Afiadenyo M., Kwofie S. K., Wilson M. D., Kusi K. A., Obiri-Yeboah D., et al. (2023). In silico screening of phytochemicals from dissotisrotundifolia against plasmodium falciparum dihydrofolate reductase. Phytomedicine Plus 3 (2), 100447. 10.1016/j.phyplu.2023.100447 - DOI
LinkOut - more resources
Full Text Sources