

Identification of an exosite at the neutrophil elastase/alpha-1-antitrypsin interface
- PMID: 39777987
- PMCID: PMC12001179
- DOI: 10.1111/febs.17387
Identification of an exosite at the neutrophil elastase/alpha-1-antitrypsin interface
Abstract
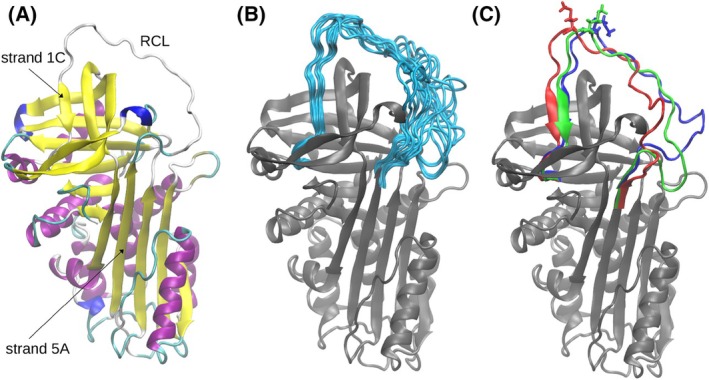
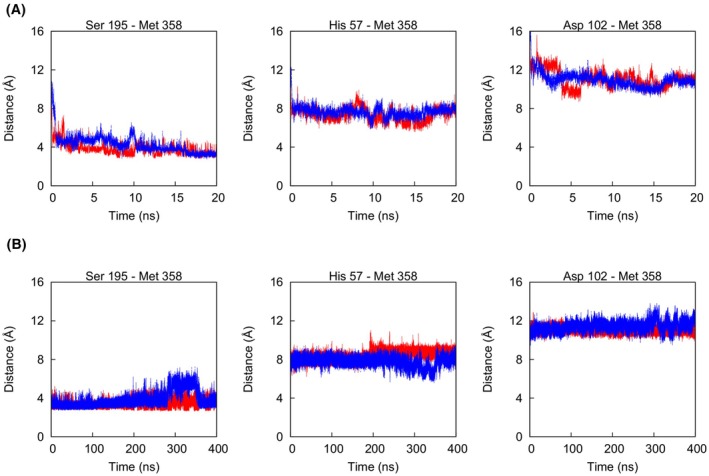
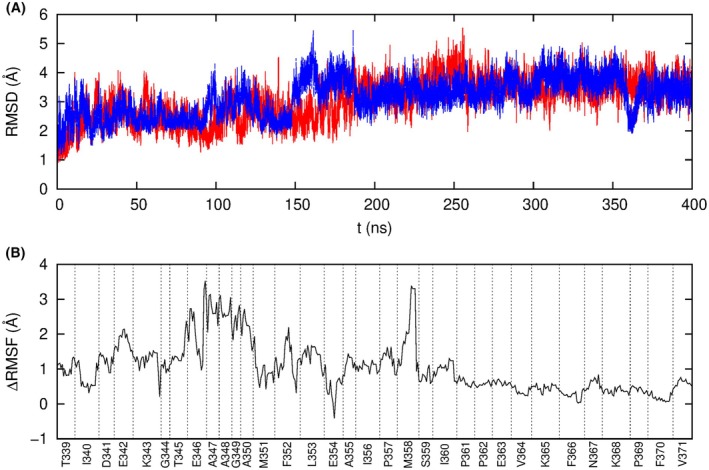
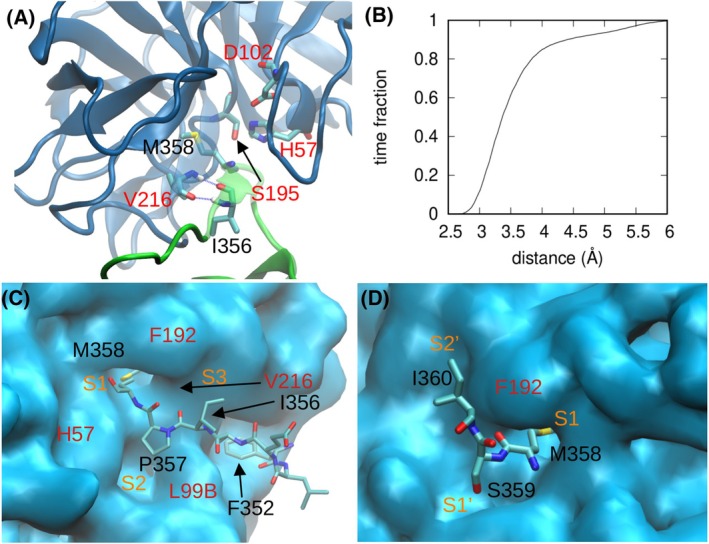
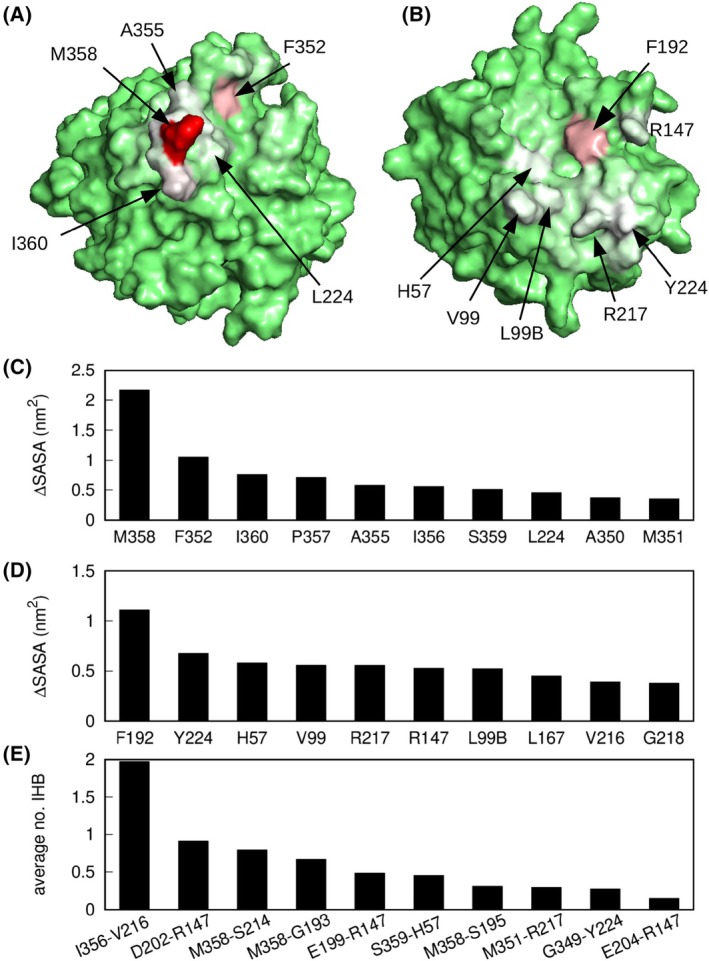
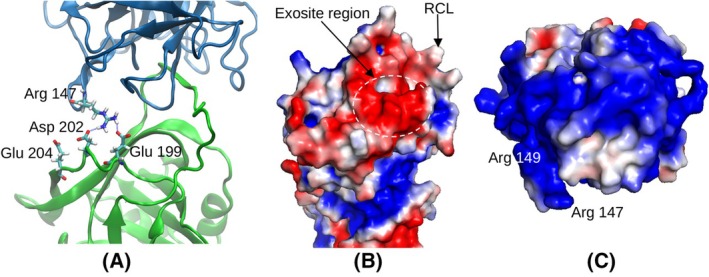
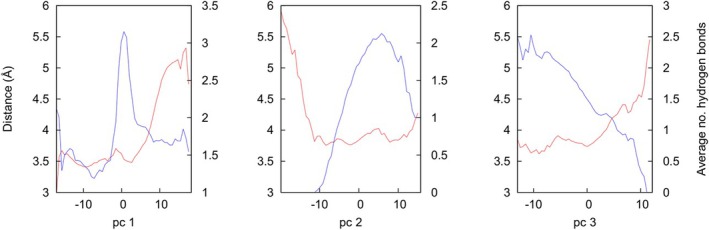
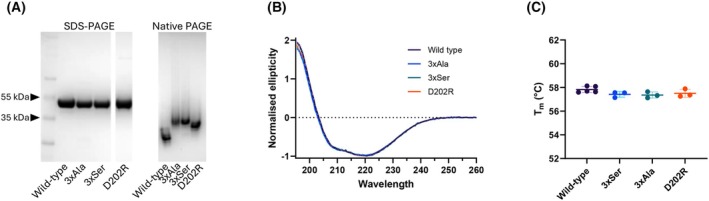
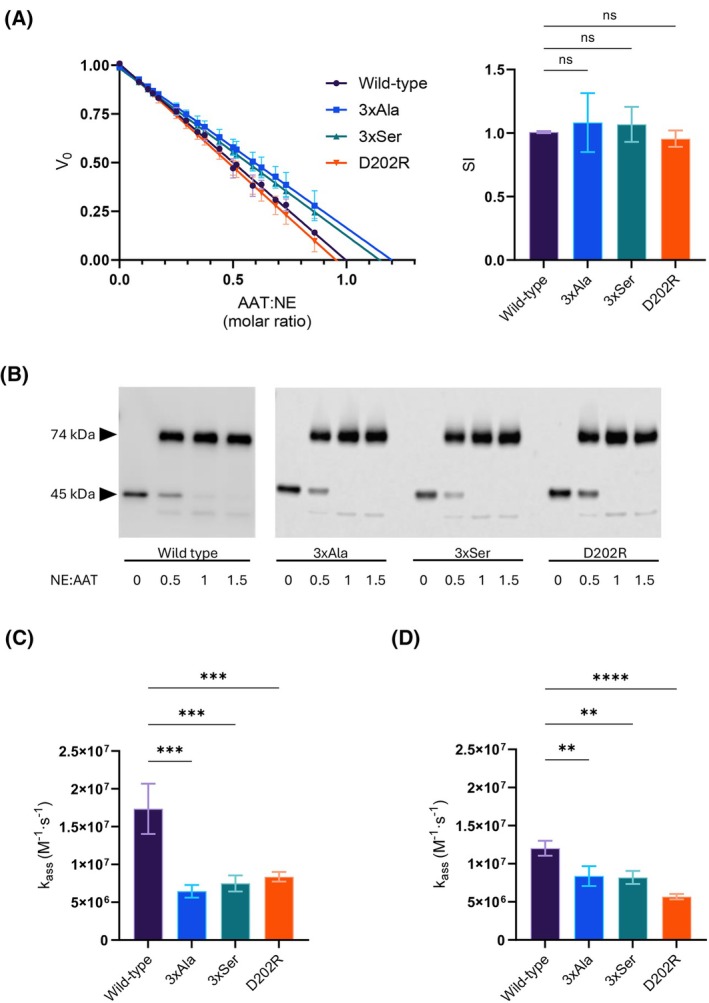
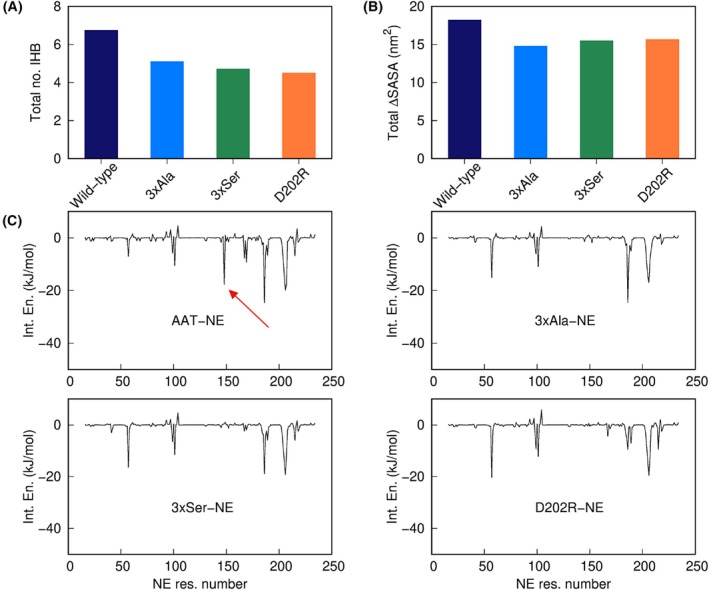
Neutrophil elastase (NE) is released by activated neutrophils during an inflammatory response and exerts proteolytic activity on elastin and other extracellular matrix components. This protease is rapidly inhibited by the plasma serine protease inhibitor alpha-1-antitrypsin (AAT), and the importance of this protective activity on lung tissue is highlighted by the development of early onset emphysema in individuals with AAT deficiency. As a serpin, AAT presents a surface-exposed reactive centre loop (RCL) whose sequence mirrors the target protease specificity. Following binding of NE in a 'Michaelis' encounter complex, cleavage of the RCL results in an irreversible complex between the two molecules. Here, the structure of the AAT-NE encounter complex was studied by molecular dynamics, mutagenesis and enzyme kinetics. Exploration of the geometry of interaction between the two molecules revealed the possibility that the interaction interface extends beyond the RCL; a persistent feature of the simulations was the interaction between a region located upstream of β-strand 4C of AAT, comprising three acidic residues (Asp202, Glu199 and Glu204), and Arg147 of NE. Mutation of the acidic residues to either alanine or serine, or a D202R substitution, resulted in a reduced rate of association between recombinant AAT and NE. Addition of salt to the buffer had little effect for these mutants but substantially reduced the rate of interaction of the wild-type protein. These data are consistent with a role for this acidic region on AAT as an exosite that contributes to an optimal interaction with its physiological protease target.
Keywords: SERPINA1; Serpins; alpha‐1‐antitrypsin deficiency; computational biology; molecular dynamics simulations; protease inhibition; protein–protein interactions.
© 2025 The Author(s). The FEBS Journal published by John Wiley & Sons Ltd on behalf of Federation of European Biochemical Societies.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- McRae B, Nakajima K, Travis J & Powers JC (1980) Studies on reactivity of human leukocyte elastase, cathepsin G, and porcine pancreatic elastase toward peptides including sequences related to the reactive site of alpha 1‐protease inhibitor (alpha 1‐antitrypsin). Biochemistry 19, 3973–3978. - PubMed
-
- Duranton J & Bieth JG (2003) Inhibition of proteinase 3 by [alpha]1‐antitrypsin in vitro predicts very fast inhibition in vivo. Am J Respir Cell Mol Biol 29, 57–61. - PubMed
-
- Greene CM, Marciniak SJ, Teckman J, Ferrarotti I, Brantly ML, Lomas DA, Stoller JK & McElvaney NG (2016) α1‐antitrypsin deficiency. Nat Rev Dis Primers 2, 16051. - PubMed
-
- Strnad P, McElvaney NG & Lomas DA (2020) Alpha1‐antitrypsin deficiency. N Engl J Med 382, 1443–1455. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
