

Benchmarking cross-species single-cell RNA-seq data integration methods: towards a cell type tree of life
- PMID: 39778870
- PMCID: PMC11707536
- DOI: 10.1093/nar/gkae1316
Benchmarking cross-species single-cell RNA-seq data integration methods: towards a cell type tree of life
Abstract
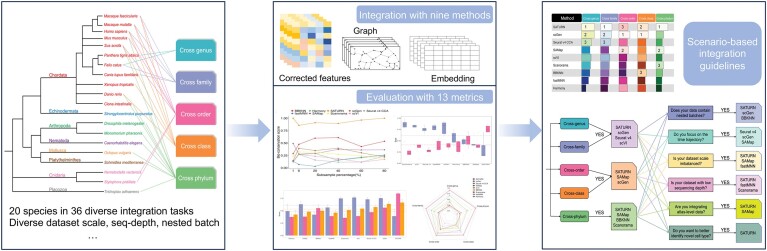
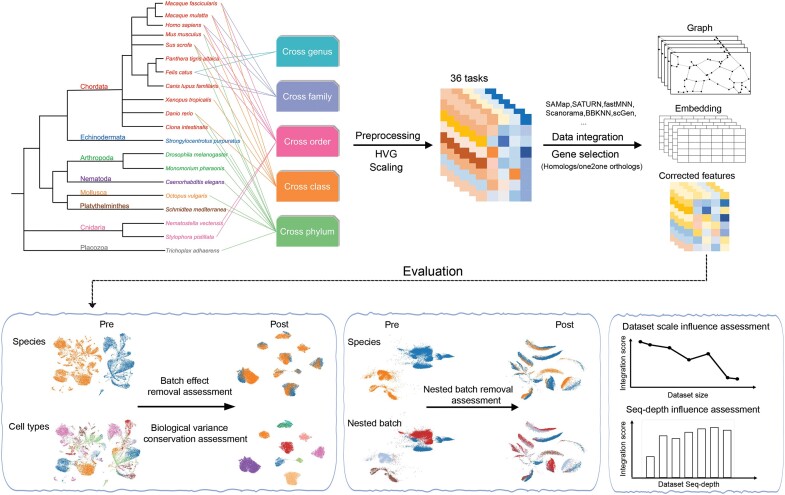
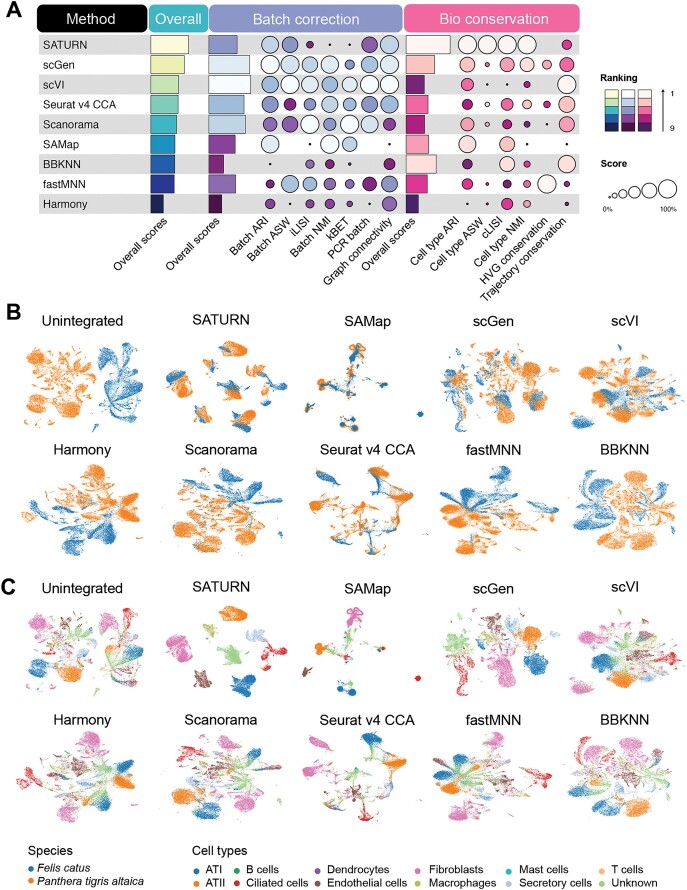
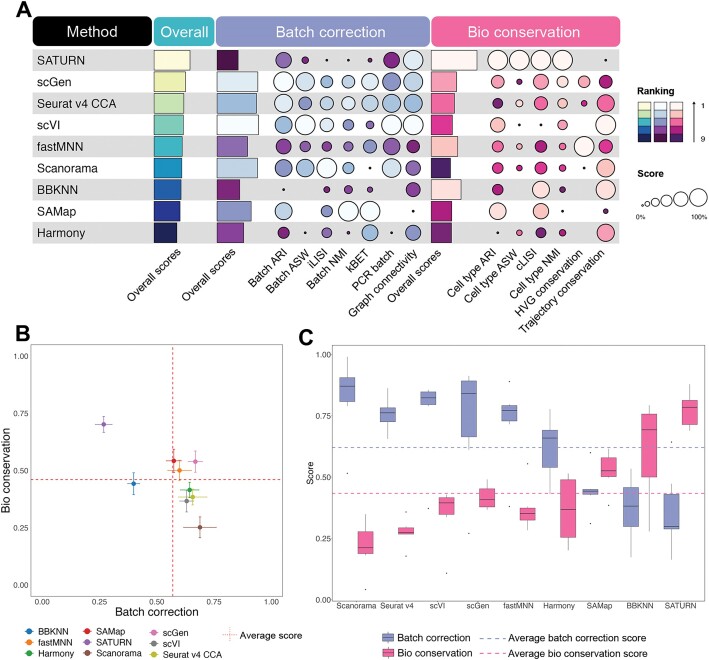
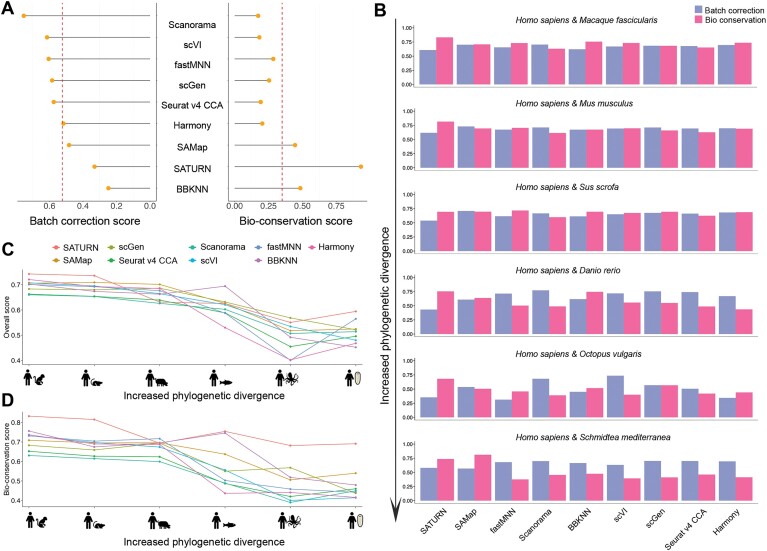
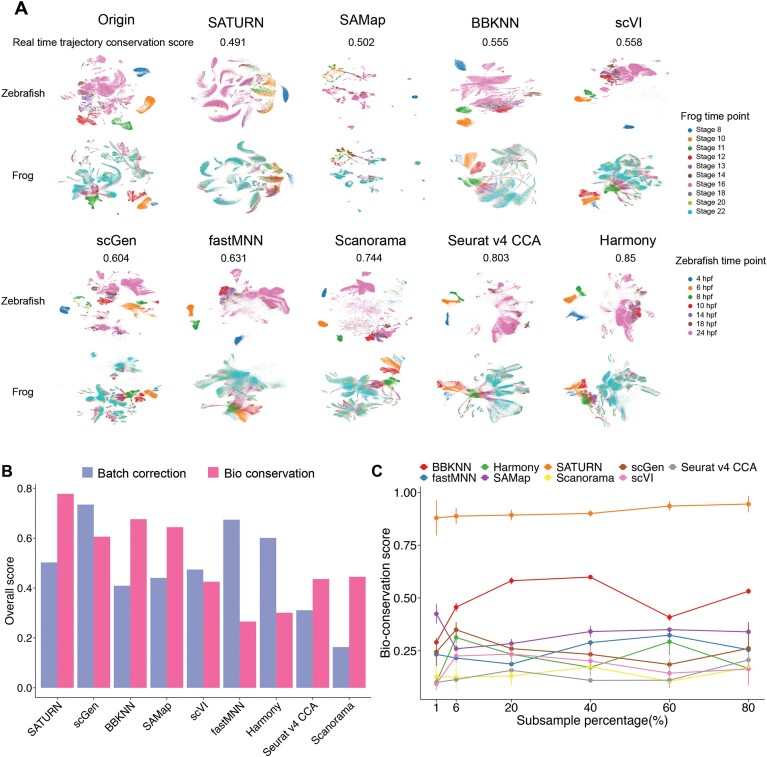
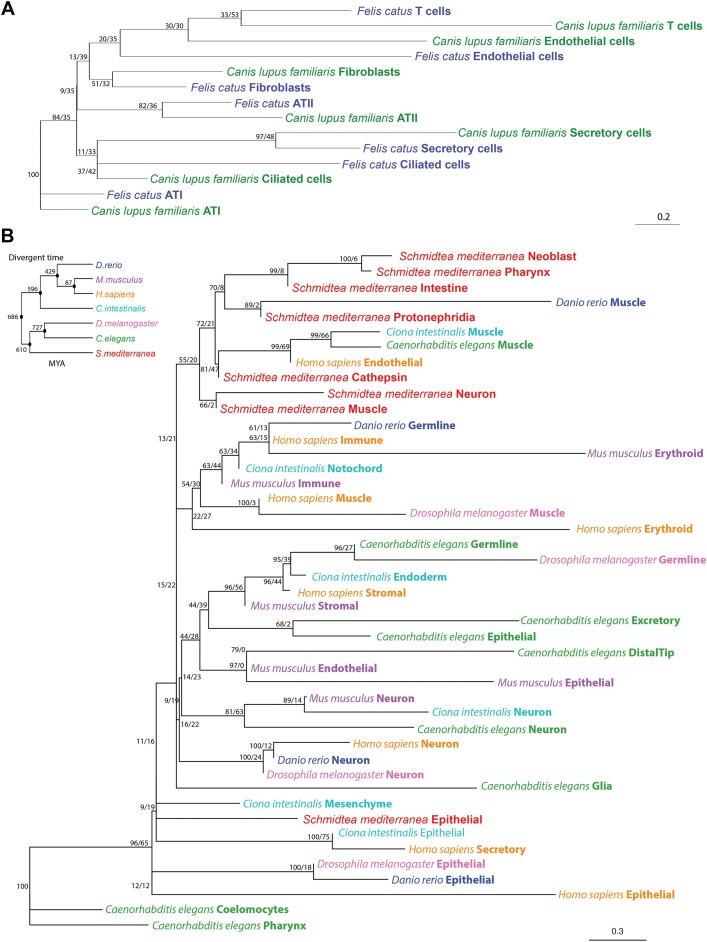
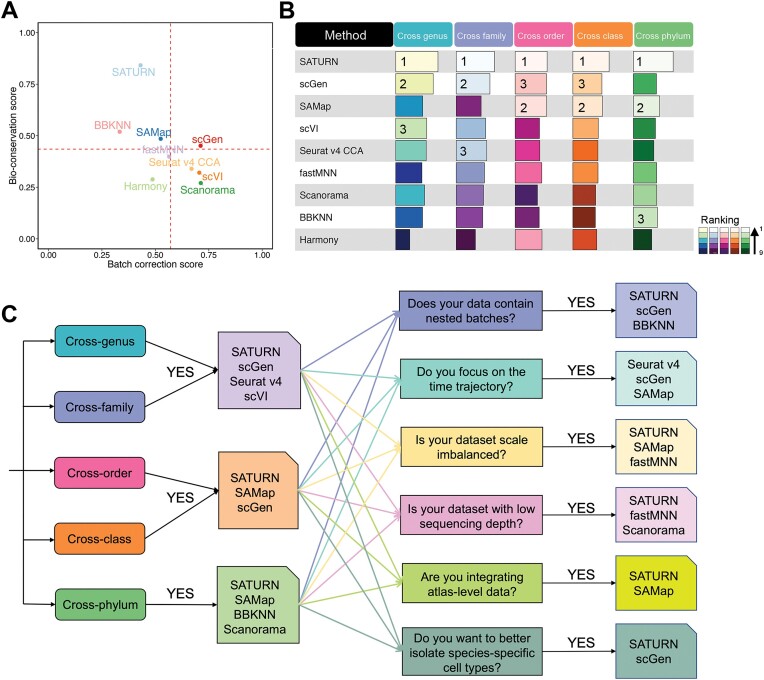
Cross-species single-cell RNA-seq data hold immense potential for unraveling cell type evolution and transferring knowledge between well-explored and less-studied species. However, challenges arise from interspecific genetic variation, batch effects stemming from experimental discrepancies and inherent individual biological differences. Here, we benchmarked nine data-integration methods across 20 species, encompassing 4.7 million cells, spanning eight phyla and the entire animal taxonomic hierarchy. Our evaluation reveals notable differences between the methods in removing batch effects and preserving biological variance across taxonomic distances. Methods that effectively leverage gene sequence information capture underlying biological variances, while generative model-based approaches excel in batch effect removal. SATURN demonstrates robust performance across diverse taxonomic levels, from cross-genus to cross-phylum, emphasizing its versatility. SAMap excels in integrating species beyond the cross-family level, especially for atlas-level cross-species integration, while scGen shines within or below the cross-class hierarchy. As a result, our analysis offers recommendations and guidelines for selecting suitable integration methods, enhancing cross-species single-cell RNA-seq analyses and advancing algorithm development.
© The Author(s) 2025. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
Similar articles
-
Benchmarking atlas-level data integration in single-cell genomics.Nat Methods. 2022 Jan;19(1):41-50. doi: 10.1038/s41592-021-01336-8. Epub 2021 Dec 23. Nat Methods. 2022. PMID: 34949812 Free PMC article.
-
XgCPred: Cell type classification using XGBoost-CNN integration and exploiting gene expression imaging in single-cell RNAseq data.Comput Biol Med. 2024 Oct;181:109066. doi: 10.1016/j.compbiomed.2024.109066. Epub 2024 Aug 24. Comput Biol Med. 2024. PMID: 39180857
-
Clustering scRNA-seq data with the cross-view collaborative information fusion strategy.Brief Bioinform. 2024 Sep 23;25(6):bbae511. doi: 10.1093/bib/bbae511. Brief Bioinform. 2024. PMID: 39402696 Free PMC article.
-
Fourteen years of cellular deconvolution: methodology, applications, technical evaluation and outstanding challenges.Nucleic Acids Res. 2024 May 22;52(9):4761-4783. doi: 10.1093/nar/gkae267. Nucleic Acids Res. 2024. PMID: 38619038 Free PMC article. Review.
-
Integration tools for scRNA-seq data and spatial transcriptomics sequencing data.Brief Funct Genomics. 2024 Jul 19;23(4):295-302. doi: 10.1093/bfgp/elae002. Brief Funct Genomics. 2024. PMID: 38267084 Review.
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
