

A foundation model of transcription across human cell types
- PMID: 39779852
- PMCID: PMC11754112
- DOI: 10.1038/s41586-024-08391-z
A foundation model of transcription across human cell types
Abstract
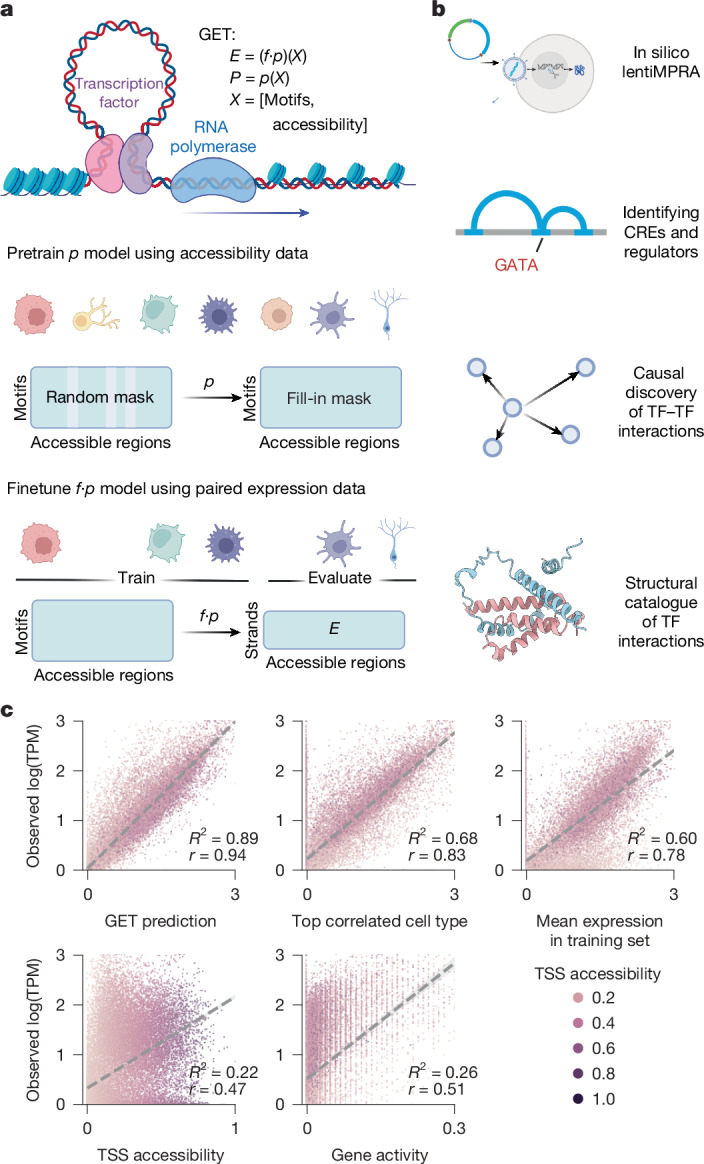
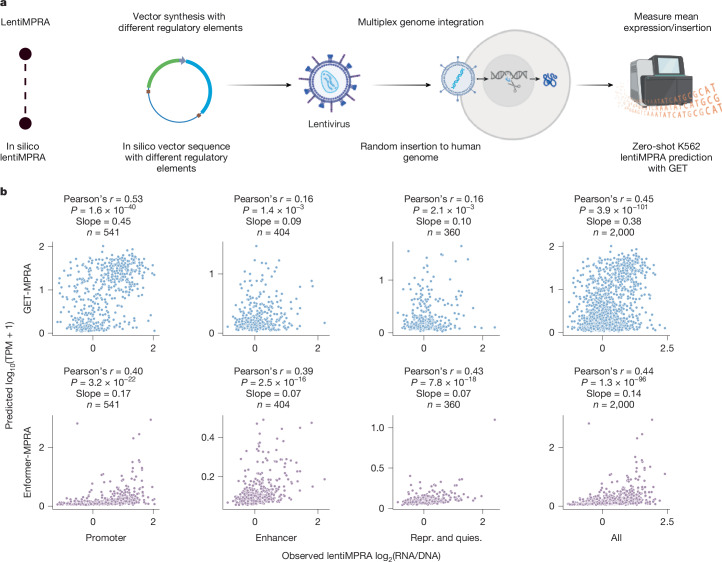
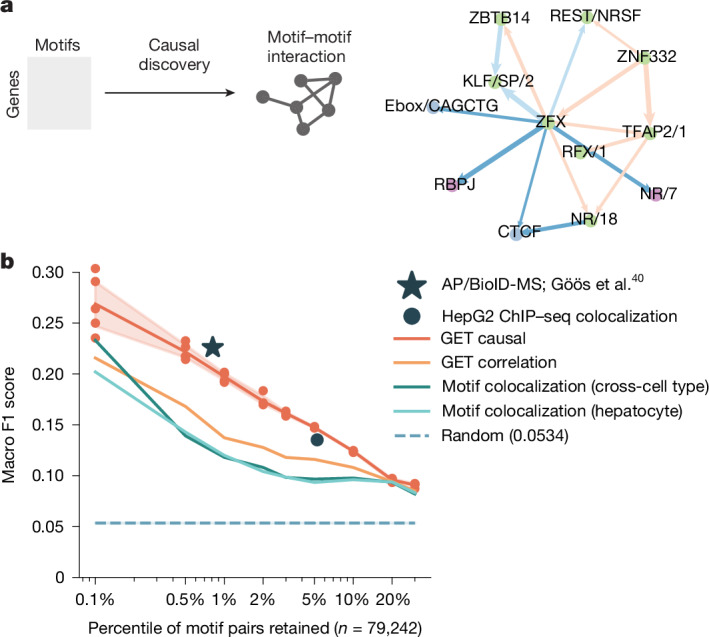
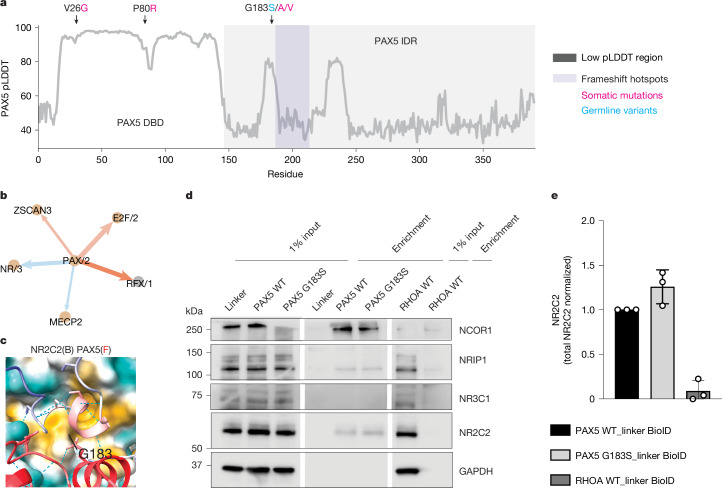
Transcriptional regulation, which involves a complex interplay between regulatory sequences and proteins, directs all biological processes. Computational models of transcription lack generalizability to accurately extrapolate to unseen cell types and conditions. Here we introduce GET (general expression transformer), an interpretable foundation model designed to uncover regulatory grammars across 213 human fetal and adult cell types1,2. Relying exclusively on chromatin accessibility data and sequence information, GET achieves experimental-level accuracy in predicting gene expression even in previously unseen cell types3. GET also shows remarkable adaptability across new sequencing platforms and assays, enabling regulatory inference across a broad range of cell types and conditions, and uncovers universal and cell-type-specific transcription factor interaction networks. We evaluated its performance in prediction of regulatory activity, inference of regulatory elements and regulators, and identification of physical interactions between transcription factors and found that it outperforms current models4 in predicting lentivirus-based massively parallel reporter assay readout5,6. In fetal erythroblasts7, we identified distal (greater than 1 Mbp) regulatory regions that were missed by previous models, and, in B cells, we identified a lymphocyte-specific transcription factor-transcription factor interaction that explains the functional significance of a leukaemia risk predisposing germline mutation8-10. In sum, we provide a generalizable and accurate model for transcription together with catalogues of gene regulation and transcription factor interactions, all with cell type specificity.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: A provisional patent with application numbers 63/486,855 and PCT/US2024/017064 has been filed by Columbia University on the use of the method developed in this work to identify gene regulatory elements and alter gene regulation and expression; X.F. and R.R. are inventors on this patent. R.R. is a founder of Genotwin and a member of the SAB of Diatech Pharmacogenomics and Flahy. None of these activities are related to the work described in this manuscript.
Figures












Update of
-
GET: a foundation model of transcription across human cell types.bioRxiv [Preprint]. 2024 Jul 3:2023.09.24.559168. doi: 10.1101/2023.09.24.559168. bioRxiv. 2024. Update in: Nature. 2025 Jan;637(8047):965-973. doi: 10.1038/s41586-024-08391-z. PMID: 39005360 Free PMC article. Updated. Preprint.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
