

A rare PRIMER cell state in plant immunity
- PMID: 39779856
- PMCID: PMC11798839
- DOI: 10.1038/s41586-024-08383-z
A rare PRIMER cell state in plant immunity
Abstract
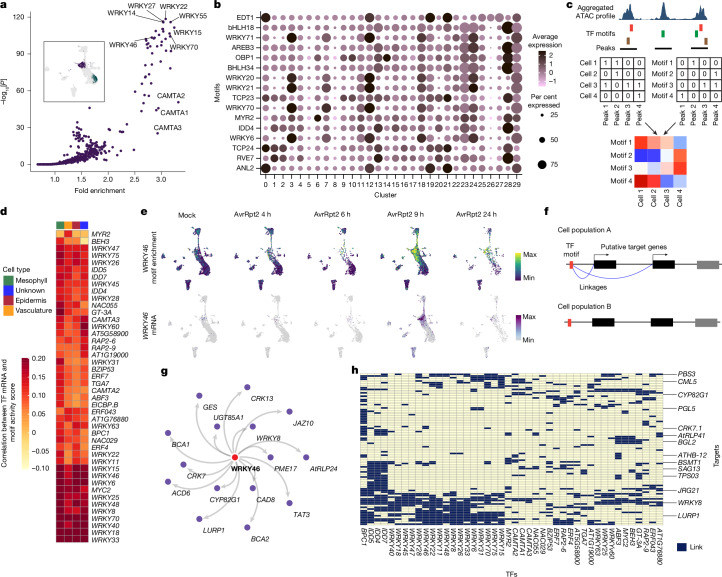
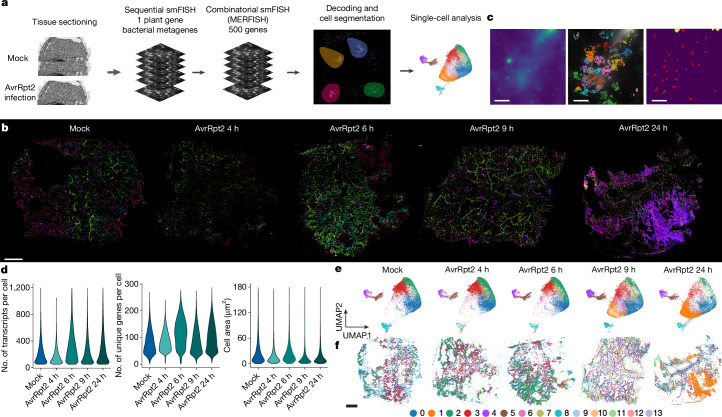
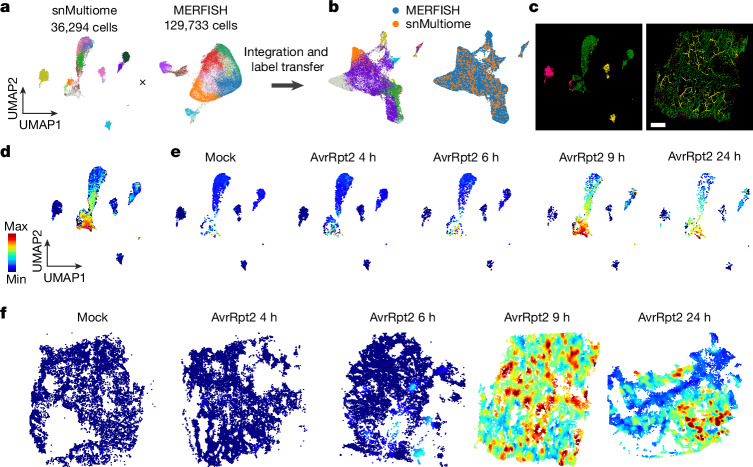
Plants lack specialized and mobile immune cells. Consequently, any cell type that encounters pathogens must mount immune responses and communicate with surrounding cells for successful defence. However, the diversity, spatial organization and function of cellular immune states in pathogen-infected plants are poorly understood1. Here we infect Arabidopsis thaliana leaves with bacterial pathogens that trigger or supress immune responses and integrate time-resolved single-cell transcriptomic, epigenomic and spatial transcriptomic data to identify cell states. We describe cell-state-specific gene-regulatory logic that involves transcription factors, putative cis-regulatory elements and target genes associated with disease and immunity. We show that a rare cell population emerges at the nexus of immune-active hotspots, which we designate as primary immune responder (PRIMER) cells. PRIMER cells have non-canonical immune signatures, exemplified by the expression and genome accessibility of a previously uncharacterized transcription factor, GT-3A, which contributes to plant immunity against bacterial pathogens. PRIMER cells are surrounded by another cell state (bystander) that activates genes for long-distance cell-to-cell immune signalling. Together, our findings suggest that interactions between these cell states propagate immune responses across the leaf. Our molecularly defined single-cell spatiotemporal atlas provides functional and regulatory insights into immune cell states in plants.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: J.R.E. serves on the scientific advisory board of Zymo Research and of Cibus. The other authors declare no competing interests.
Figures











References
-
- Tang, B., Feng, L., Hulin, M. T., Ding, P. & Ma, W. Cell-type-specific responses to fungal infection in plants revealed by single-cell transcriptomics. Cell Host Microbe10.1016/j.chom.2023.08.019 (2023). - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
