

Enterobactin inhibits microbiota-dependent activation of AhR to promote bacterial sepsis in mice
- PMID: 39779878
- PMCID: PMC11905502
- DOI: 10.1038/s41564-024-01882-9
Enterobactin inhibits microbiota-dependent activation of AhR to promote bacterial sepsis in mice
Abstract
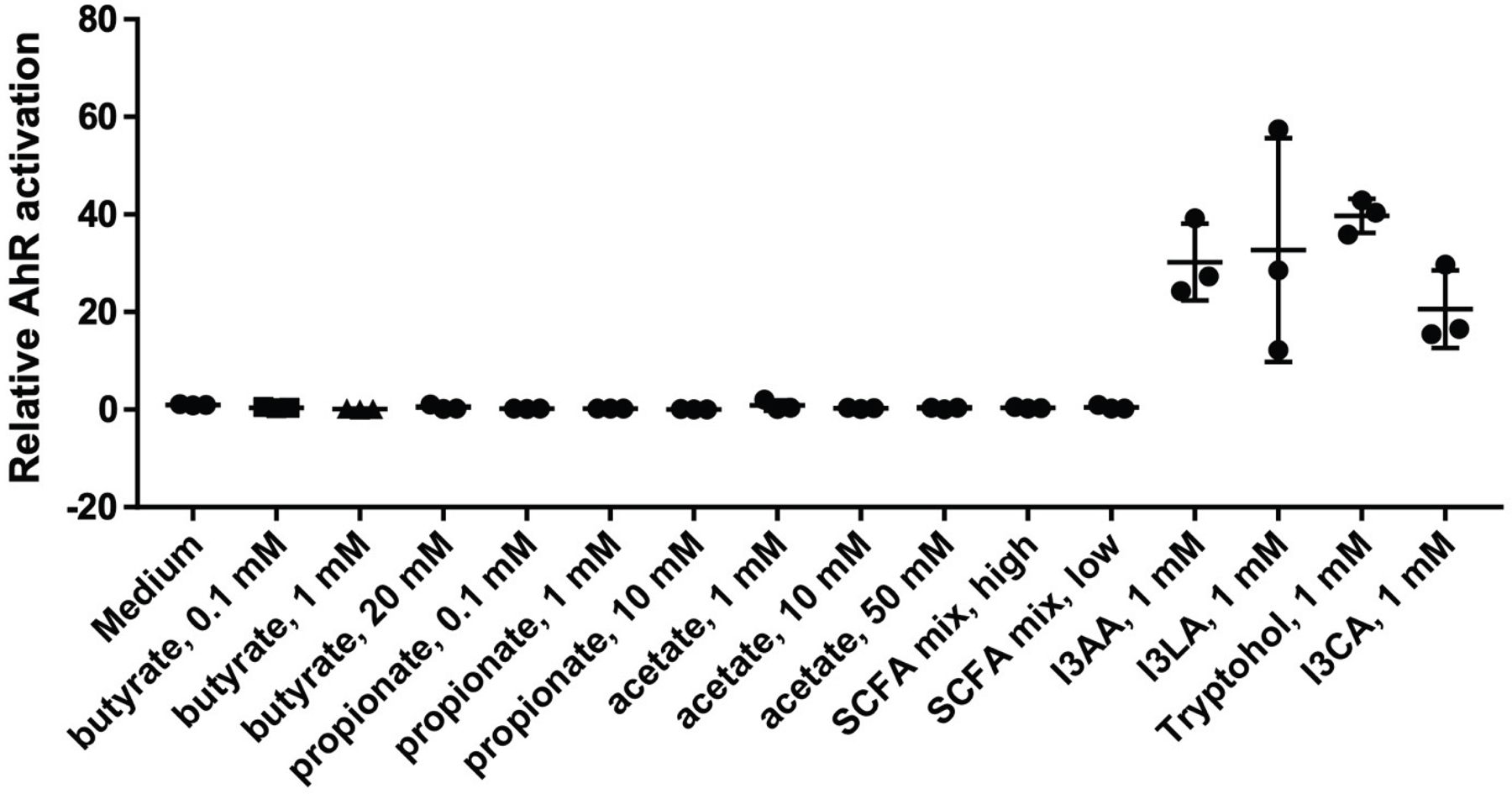
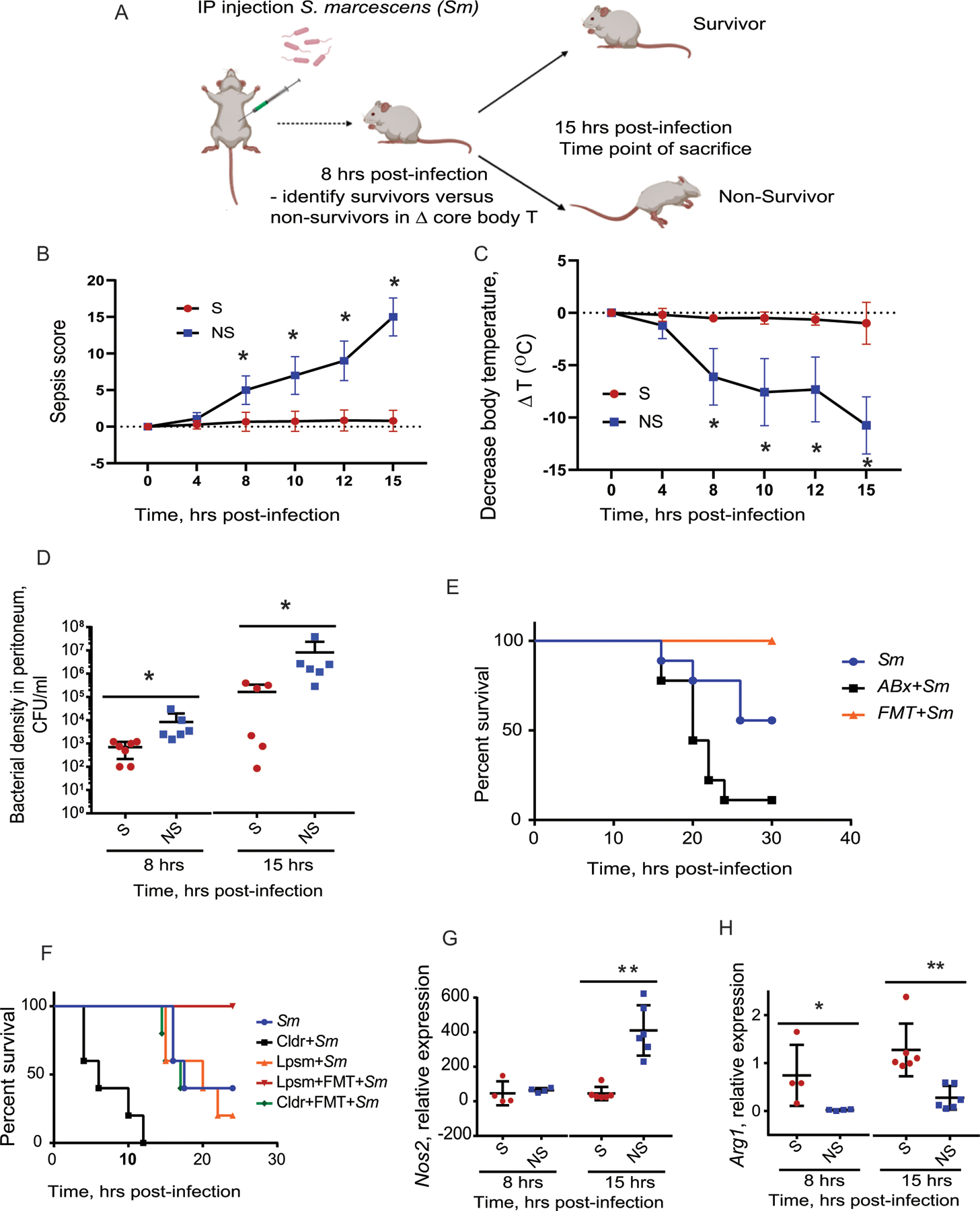
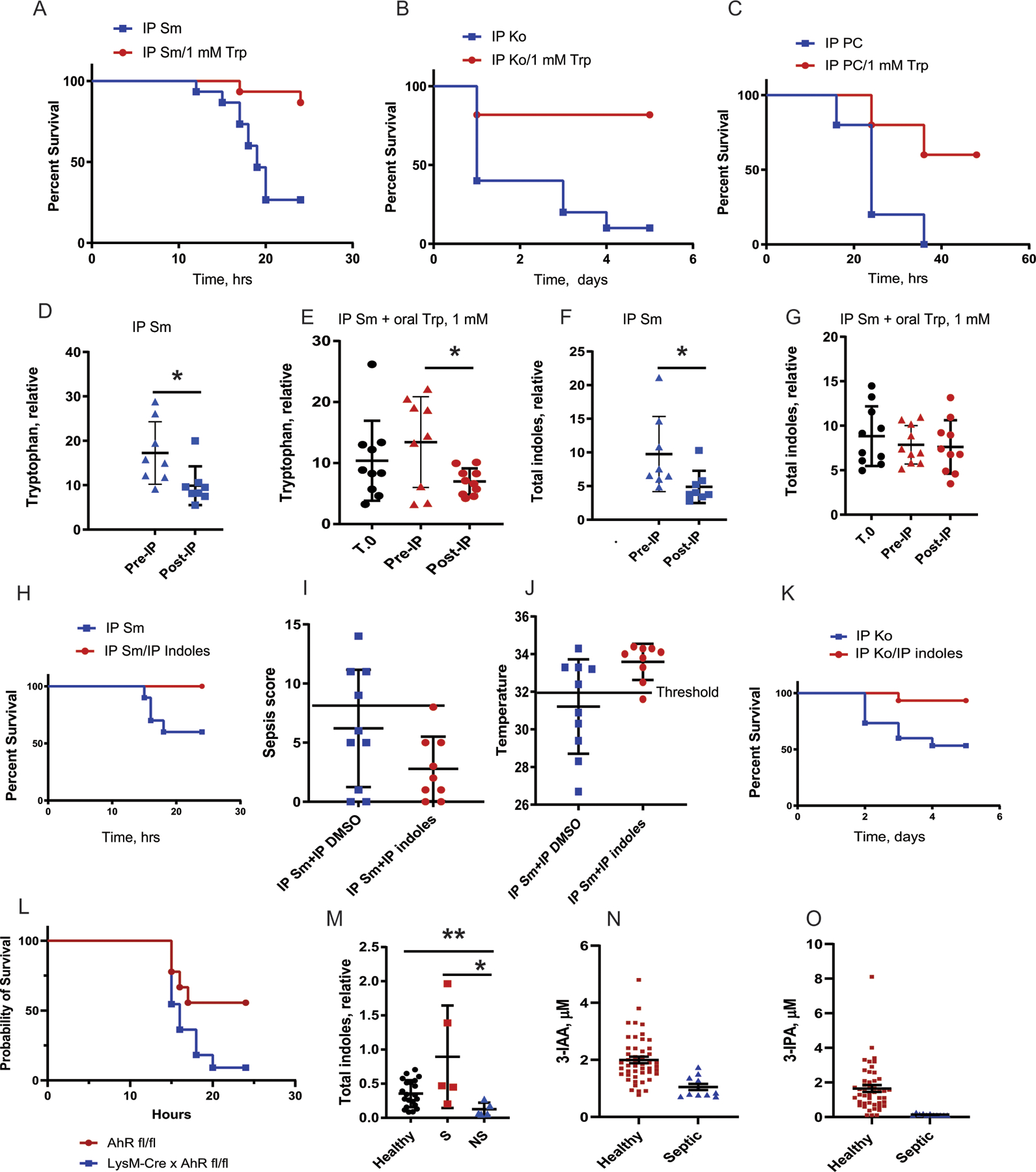
Sepsis is a major cause of morbidity and mortality, but our understanding of the mechanisms underlying survival or susceptibility is limited. Here, as pathogens often subvert host defence mechanisms, we hypothesized that this might influence the outcome of sepsis. We used microbiota analysis, faecal microbiota transplantation, antibiotic treatment and caecal metabolite analysis to show that gut-microbiota-derived tryptophan metabolites including indoles increased host survival in a mouse model of Serratia marcescens sepsis. Infection in macrophage-specific aryl hydrocarbon receptor (AhR) knockout mice revealed that AhR activation induced transcriptional reprogramming in macrophages and increased bacterial clearance and host survival. However, culture supernatants from multiple bacterial pathogens inhibited AhR activation in vitro. We showed that the secreted siderophore, enterobactin, inhibited AhR activation in vitro and increased sepsis mortality in vivo. By contrast, oral or systemic tryptophan supplementation increased survival. These findings show that sepsis survival depends upon the interplay between pathogen inhibition and the activation of AhR by a microbiota-derived metabolite.
© 2025. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
Competing interests: The authors declare nocompeting interests.
Figures













References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
