

The modern use of hydroxyurea for children with sickle cell anemia
- PMID: 39781621
- PMCID: PMC12050929
- DOI: 10.3324/haematol.2023.284633
The modern use of hydroxyurea for children with sickle cell anemia
Abstract
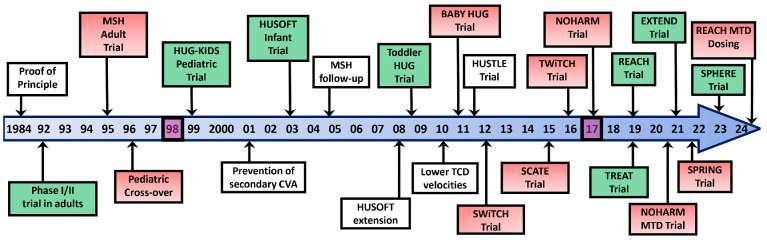
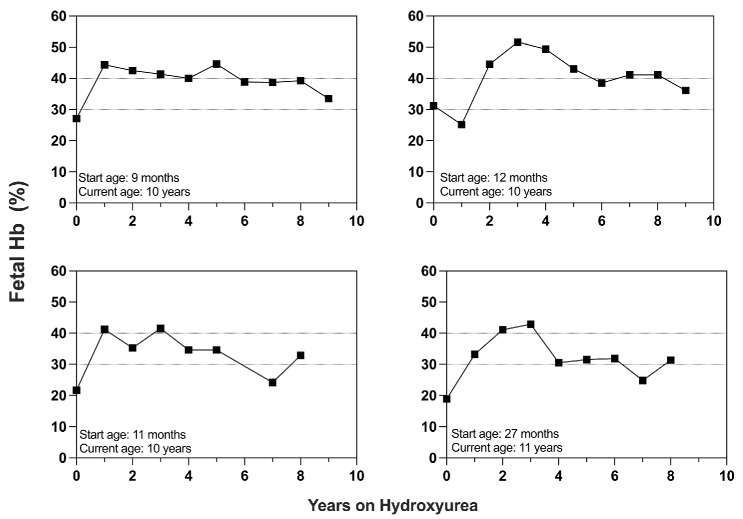
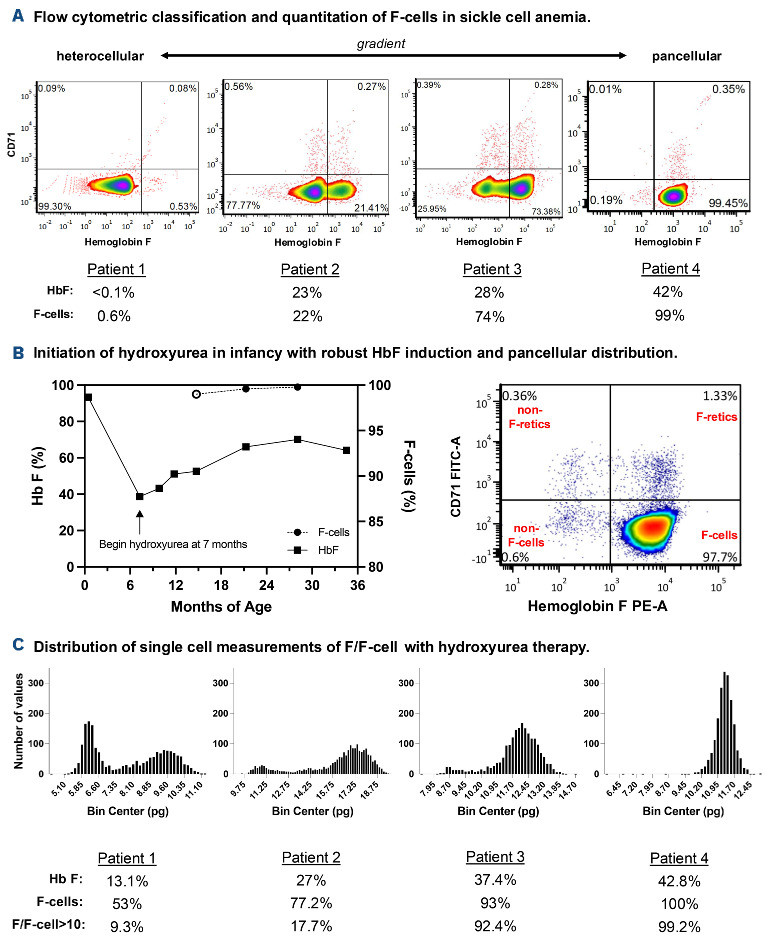
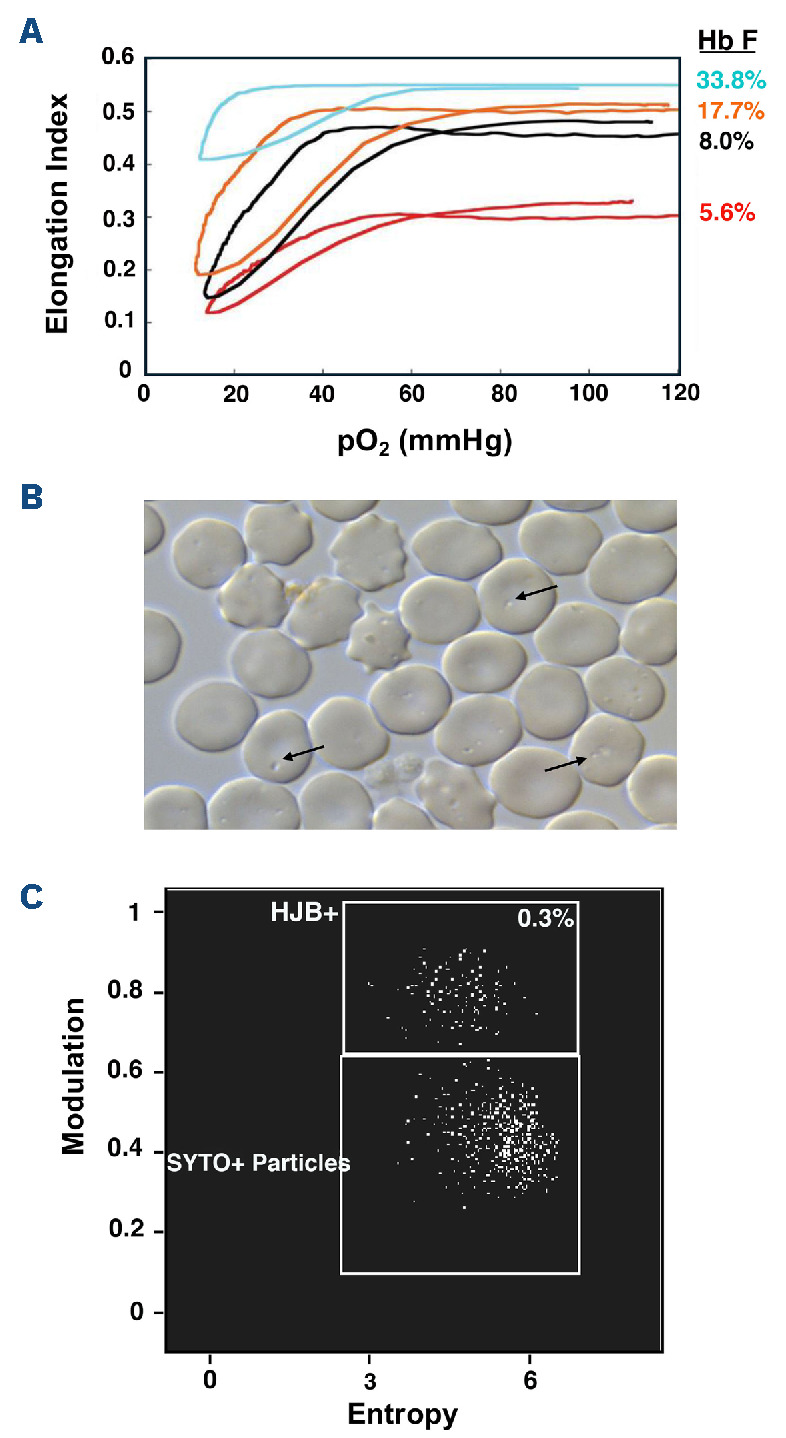
Over the past 40 years, the introduction and refinement of hydroxyurea therapy has led to remarkable progress in the care of individuals with sickle cell anemia (SCA). From initial small proof-of-principle studies to multicenter phase III controlled clinical trials and then numerous open-label studies, the consistent benefits of once-daily oral hydroxyurea have been demonstrated across the lifespan. Elevated fetal hemoglobin (HbF) serves as the most important treatment response, as HbF delays sickle hemoglobin polymerization and reduces erythrocyte sickling. Increased amounts of HbF, especially when distributed across the majority of erythrocytes, improve clinical outcomes by reducing hemolytic anemia and preventing vaso-occlusion, thereby ameliorating both acute and chronic - and overt and covert - complications. Additional benefits of hydroxyurea beyond HbF induction include lower neutrophil and platelet counts, reduced inflammation, and improved rheology. Toxicities of hydroxyurea in SCA are typically mild and predictable; modest cytopenia is expected and actually therapeutic, while occasional gastrointestinal and cutaneous manifestations are well-tolerated. Long-term risks of hydroxyurea for SCA are mainly theoretical but require ongoing surveillance. Accordingly, hydroxyurea should be initiated as part of standard of care, ideally in the first year of life. Proper dosing of hydroxyurea is critical, aiming through stepwise dose escalation to achieve modest but safe myelosuppression, with periodic adjustments for weight gain. Precision dosing using pharmacokinetics may facilitate optimal dosing without frequent dose adjustments. Although transformative and even curative therapies for SCA are emerging, hydroxyurea is the only available and accessible disease-modifying treatment that can address the global burden of disease, especially in low-resource settings within sub-Saharan Africa.
Figures
References
-
- Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. . Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA. 2014;312(1):1033-1048. - PubMed
-
- Watson J. The significance of the paucity of sickle cells in newborn Negro infants. Am J Med Sci. 1948;215(4):419-423. - PubMed
-
- Perrine RP, Pembrey ME, John P, Perrine S, Shoup F. Natural history of sickle cell anemia in Saudi Arabs. A study of 270 subjects. Ann Intern Med. 1978;88(1):1-6. - PubMed
-
- Jacob GF, Raper AB. Hereditary persistence of foetal haemoglobin production, and its interaction with the sickle cell trait. Br J Haematol. 1958;4(2):138-149. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
