

De novo lipogenesis protects dormant breast cancer cells from ferroptosis and promotes metastasis
- PMID: 39787900
- PMCID: PMC11764609
- DOI: 10.1016/j.redox.2024.103480
De novo lipogenesis protects dormant breast cancer cells from ferroptosis and promotes metastasis
Abstract
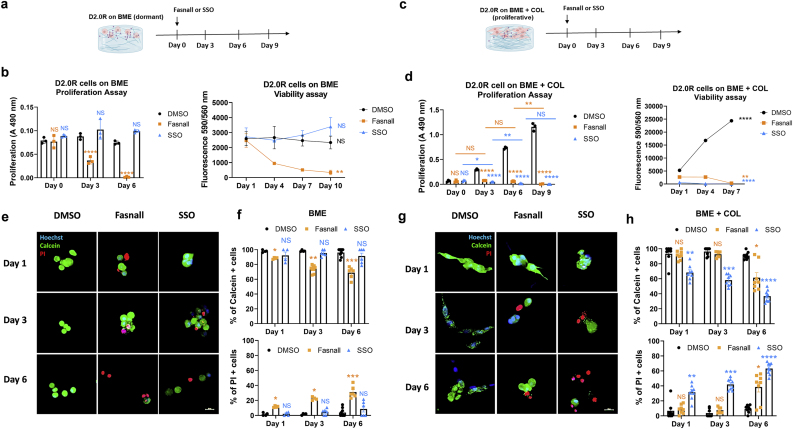
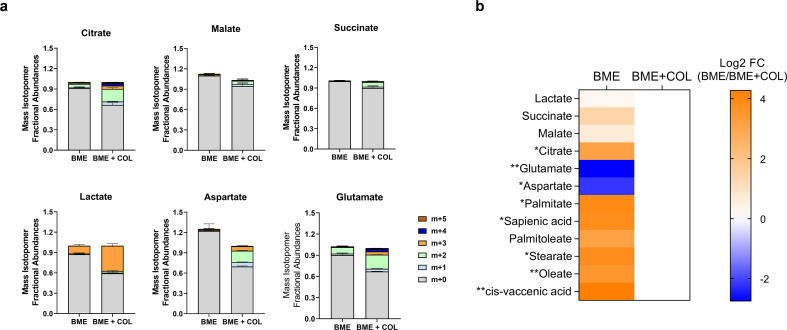
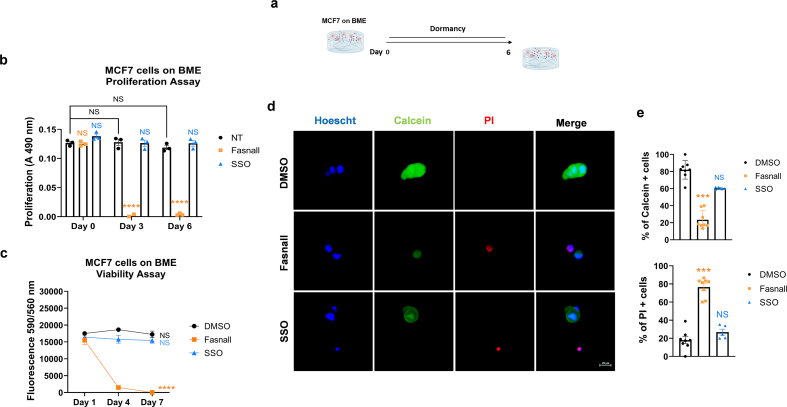
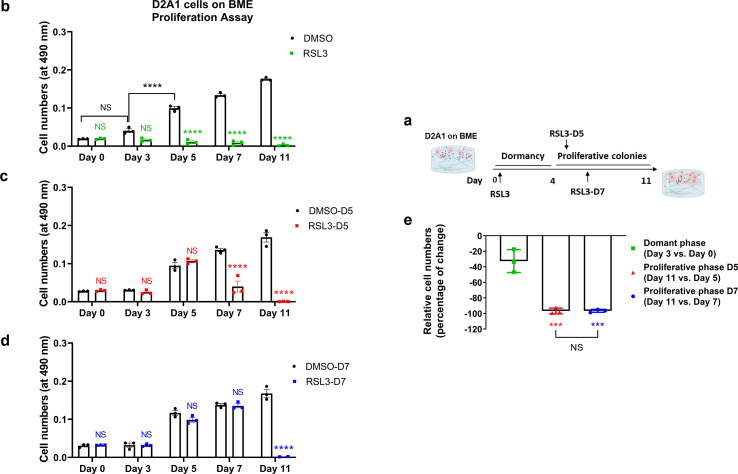
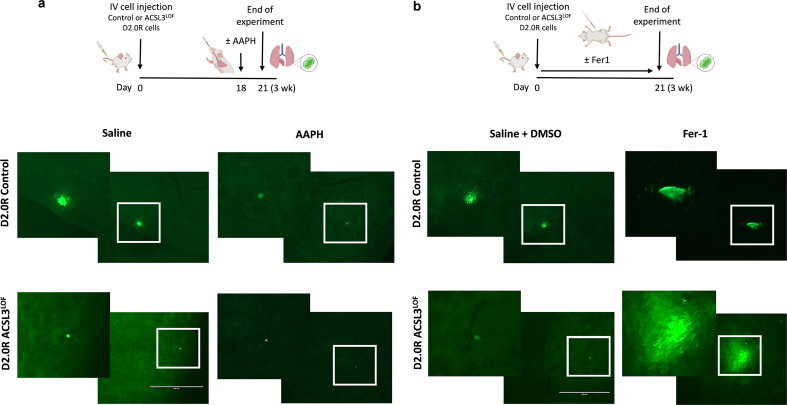
Dormant disseminated tumor cells (DTCs) remain viable for years to decades before establishing a clinically overt metastatic lesion. DTCs are known to be highly resilient and able to overcome the multiple biological hurdles imposed along the metastatic cascade. However, the specific metabolic adaptations of dormant DTCs remain to be elucidated. Here, we reveal that dormant DTCs upregulate de novo lipogenesis and favor the activation and incorporation of monounsaturated fatty acids (MUFAs) to their cellular membranes through the activation of acyl-coenzyme A synthetase long-chain family member 3 (ACSL3). Pharmacologic inhibition of de novo lipogenesis or genetic knockdown of ACSL3 results in lipid peroxidation and non-apoptotic cell death through ferroptosis. Clinically, ACSL3 was found to be overexpressed in quiescent DTCs in the lymph nodes of breast cancer patients and to significantly correlate with shorter disease-free and overall survival. Our work provides new insights into the molecular mechanisms enabling the survival of dormant DTCs and supports the use of de novo lipogenesis inhibitors to prevent breast cancer metastasis.
Keywords: Breast cancer; Ferroptosis; Lipid metabolism; Lipid peroxidation; Metastasis; Monounsaturated fatty acids activation; Tumor cell dormancy.
Copyright © 2024 The Authors. Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
Declaration of competing interest The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: S.-M.F. has received funding from BlackBelt Therapeutics, Gilead and Alesta Therapeutics, is in the advisory board of Alesta Therapeutics, has consulted for Fund+ and Droia Ventures and is in the advisory board of several Cell Press Journals. All other authors have no conflicts of interest to declare.
Figures















References
-
- Metzger-Filho O., Sun Z., Viale G., Price K.N., Crivellari D., Snyder R.D., Gelber R.D., Castiglione-Gertsch M., Coates A.S., Goldhirsch A., Cardoso F. Patterns of Recurrence and outcome according to breast cancer subtypes in lymph node-negative disease: results from international breast cancer study group trials VIII and IX. J. Clin. Oncol. 2013;31:3083–3090. doi: 10.1200/JCO.2012.46.1574. - DOI - PMC - PubMed
-
- Nobre A.R., Dalla E., Yang J., Huang X., Wullkopf L., Risson E., Razghandi P., Anton M.L., Zheng W., Seoane J.A., Curtis C., Kenigsberg E., Wang J., Aguirre-Ghiso J.A. ZFP281 drives a mesenchymal-like dormancy program in early disseminated breast cancer cells that prevents metastatic outgrowth in the lung. Nat. Cancer. 2022;3:1165–1180. doi: 10.1038/S43018-022-00424-8. - DOI - PubMed
-
- Harper K.L., Sosa M.S., Entenberg D., Hosseini H., Cheung J.F., Nobre R., Avivar-Valderas A., Nagi C., Girnius N., Davis R.J., Farias E.F., Condeelis J., Klein C.A., Aguirre-Ghiso J.A. Mechanism of early dissemination and metastasis in Her2+ mammary cancer. Nature. 2016;540:588–592. doi: 10.1038/NATURE20609. - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
