

Excited state dynamics of azanaphthalenes reveal opportunities for the rational design of photoactive molecules
- PMID: 39789245
- PMCID: PMC11717923
- DOI: 10.1038/s42004-024-01403-z
Excited state dynamics of azanaphthalenes reveal opportunities for the rational design of photoactive molecules
Abstract
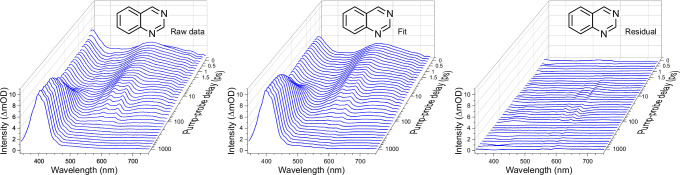
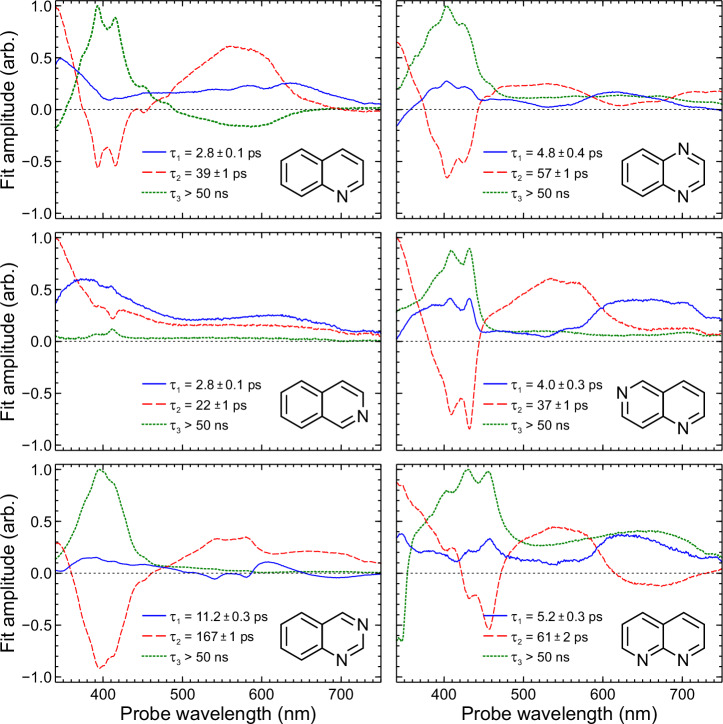
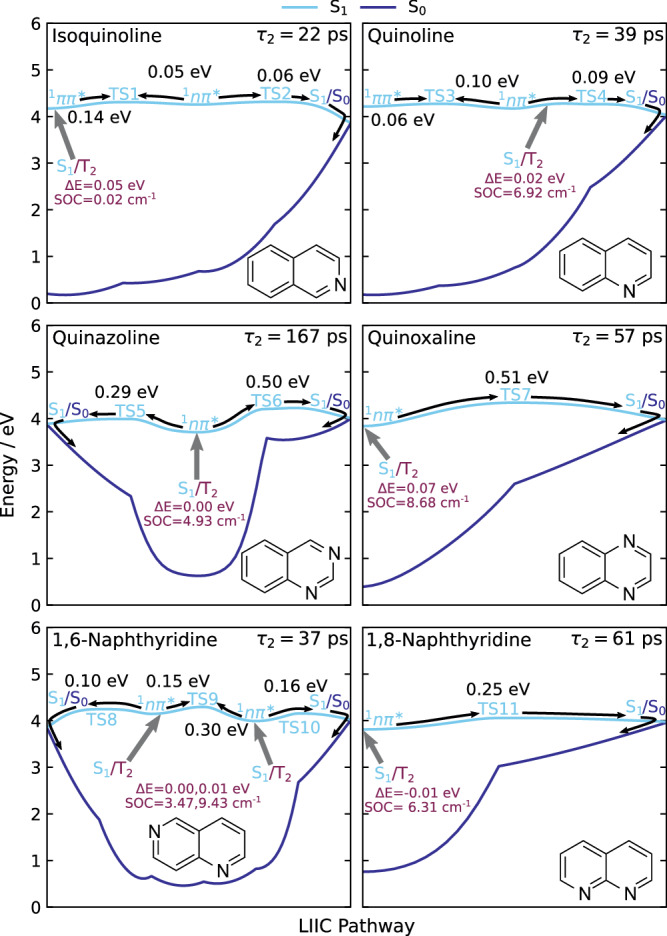
Various photoactive molecules contain motifs built on aza-aromatic heterocycles, although a detailed understanding of the excited state photophysics and photochemistry in such systems is not fully developed. To help address this issue, the non-adiabatic dynamics operating in azanaphthalenes under hexane solvation was studied following 267 nm excitation using ultrafast transient absorption spectroscopy. Specifically, the species quinoline, isoquinoline, quinazoline, quinoxaline, 1,6-naphthyridine, and 1,8-naphthyridine were investigated, providing a systematic variation in the relative positioning of nitrogen heteroatom centres within a bicyclic aromatic structure. Our results indicate considerable differences in excited state lifetimes, and in the propensity for intersystem crossing vs internal conversion across the molecular series. The overall pattern of behaviour can be explained in terms of potential energy barriers and spin-orbit coupling effects, as demonstrated by extensive quantum chemistry calculations undertaken at the SCS-ADC(2) level of theory. The fact that quantum chemistry calculations can achieve such detailed and nuanced agreement with experimental data across a full set of six molecules exhibiting subtle variations in their composition provides an excellent example of the current state-of-the-art and is indicative of future opportunities for rational design of photoactive molecules.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures





References
-
- Werner, E. R., Blau, N. & Thöny, B. Tetrahydrobiopterin: biochemistry and pathophysiology. Biochem. J.438, 397–414 (2011). - PubMed
-
- Hunger, K. & Schmidt, M. U. Industrial Organic Pigments (Wiley-VCH, 2018).
-
- Kalaria, P. N., Karad, S. C. & Raval, D. K. A review on diverse heterocyclic compounds as the privileged scaffolds in antimalarial drug discovery. Eur. J. Med. Chem.158, 917–936 (2018). - PubMed
Grants and funding
- EP/P001459/1/RCUK | Engineering and Physical Sciences Research Council (EPSRC)
- EP/T021675/1/RCUK | Engineering and Physical Sciences Research Council (EPSRC)
- EP/V006746/1/RCUK | Engineering and Physical Sciences Research Council (EPSRC)
- EP/V006819/1/RCUK | Engineering and Physical Sciences Research Council (EPSRC)
- EP/V006819/2/RCUK | Engineering and Physical Sciences Research Council (EPSRC)
- EP/V049240/2/RCUK | Engineering and Physical Sciences Research Council (EPSRC)
- EP/X026698/1/RCUK | Engineering and Physical Sciences Research Council (EPSRC)
- EP/X026973/1/RCUK | Engineering and Physical Sciences Research Council (EPSRC)
- DE-SC0020276/U.S. Department of Energy (DOE)
- RPG-2020-208/Leverhulme Trust
LinkOut - more resources
Full Text Sources
