

Single-cell Rapid Capture Hybridization sequencing reliably detects isoform usage and coding mutations in targeted genes
- PMID: 39794120
- PMCID: PMC12047256
- DOI: 10.1101/gr.279322.124
Single-cell Rapid Capture Hybridization sequencing reliably detects isoform usage and coding mutations in targeted genes
Abstract
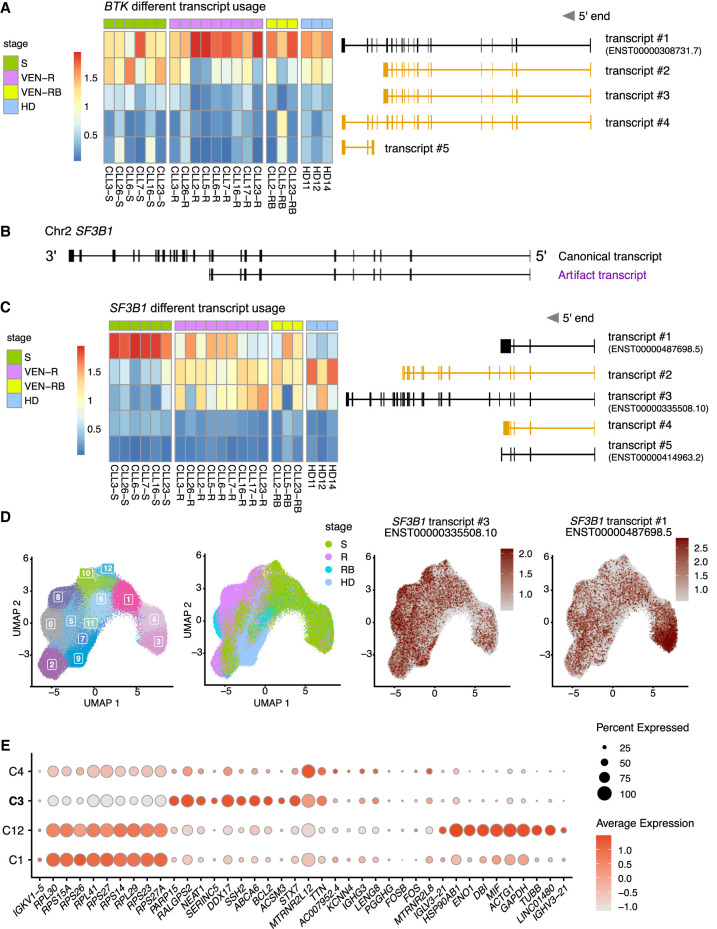
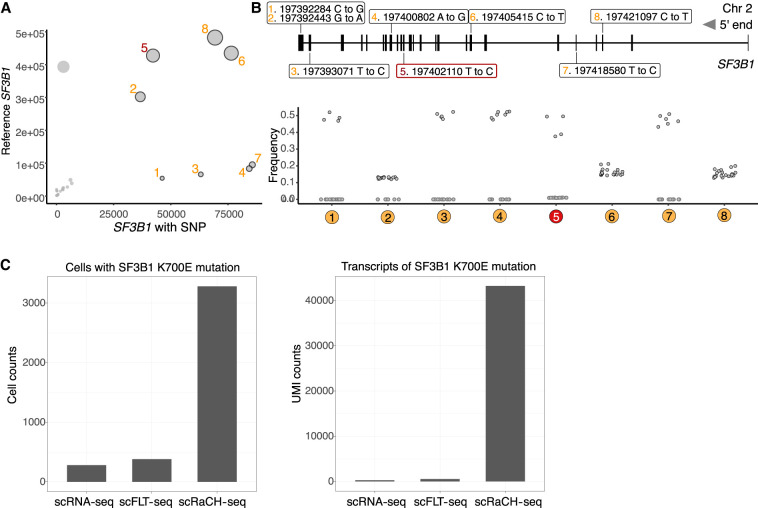
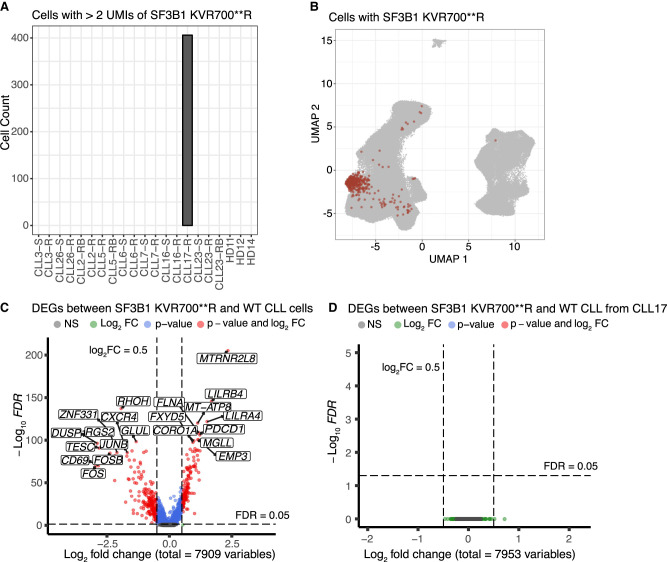
Single-cell long-read sequencing has transformed our understanding of isoform usage and the mutation heterogeneity between cells. Despite unbiased in-depth analysis, the low sequencing throughput often results in insufficient read coverage, thereby limiting our ability to perform mutation calling for specific genes. Here, we developed a single-cell Rapid Capture Hybridization sequencing (scRaCH-seq) method that demonstrates high specificity and efficiency in capturing targeted transcripts using long-read sequencing, allowing an in-depth analysis of mutation status and transcript usage for genes of interest. The method includes creating a probe panel for transcript capture, using barcoded primers for pooling and efficient sequencing via Oxford Nanopore Technologies platforms. scRaCH-seq is applicable to stored and indexed single-cell cDNA, which allows analysis to be combined with existing short-read RNA-seq data sets. In our investigation of BTK and SF3B1 genes in samples from patients with chronic lymphocytic leukemia (CLL), we detect SF3B1 isoforms and mutations with high sensitivity. Integration with short-read single-cell RNA sequencing (scRNA-seq) data reveals significant gene expression differences in SF3B1-mutated CLL cells, although it does not impact the sensitivity of the anticancer drug venetoclax. scRaCH-seq's capability to study long-read transcripts of multiple genes makes it a powerful tool for single-cell genomics.
© 2025 Peng et al.; Published by Cold Spring Harbor Laboratory Press.
Figures



References
-
- Cortés-López M, Chamely P, Hawkins AG, Stanley RF, Swett AD, Ganesan S, Mouhieddine TH, Dai X, Kluegel L, Chen C, et al. 2023. Single-cell multi-omics defines the cell-type-specific impact of splicing aberrations in human hematopoietic clonal outgrowths. Cell Stem Cell 30: 1262–1281.e8. 10.1016/j.stem.2023.07.012 - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources