

Ecological dynamics of Enterobacteriaceae in the human gut microbiome across global populations
- PMID: 39794474
- PMCID: PMC11790488
- DOI: 10.1038/s41564-024-01912-6
Ecological dynamics of Enterobacteriaceae in the human gut microbiome across global populations
Abstract
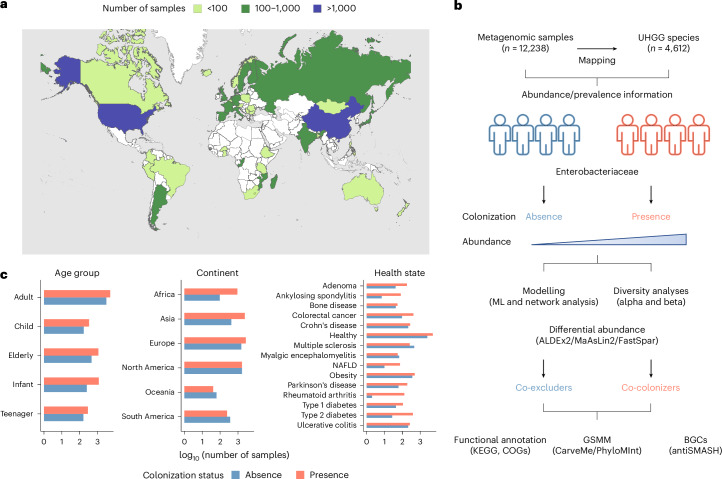
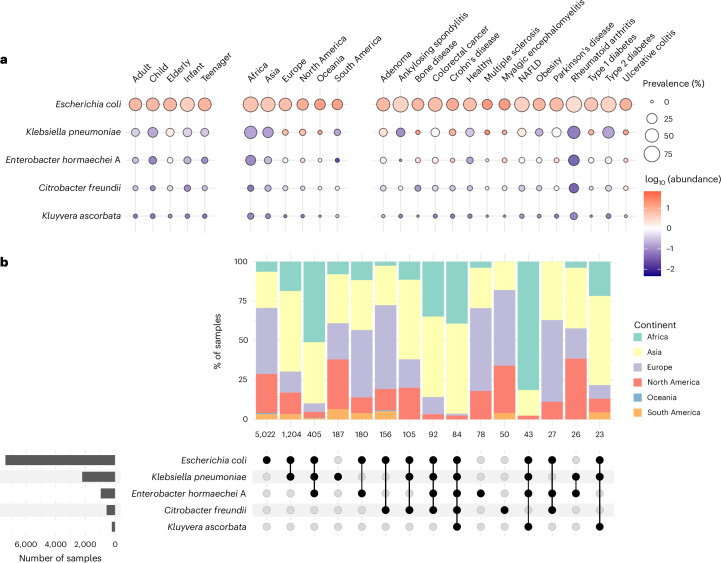
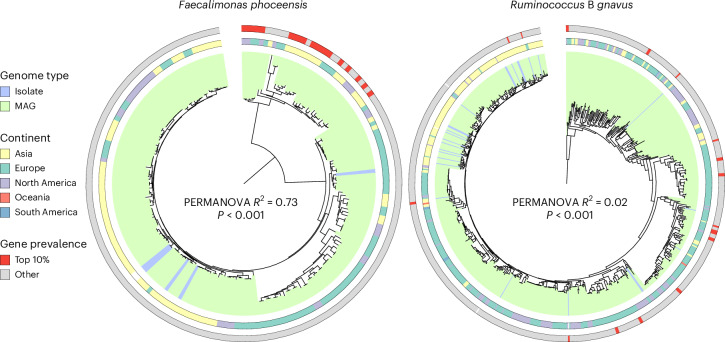
Gut bacteria from the Enterobacteriaceae family are a major cause of opportunistic infections worldwide. Given their prevalence among healthy human gut microbiomes, interspecies interactions may play a role in modulating infection resistance. Here we uncover global ecological patterns linked to Enterobacteriaceae colonization and abundance by leveraging a large-scale dataset of 12,238 public human gut metagenomes spanning 45 countries. Machine learning analyses identified a robust gut microbiome signature associated with Enterobacteriaceae colonization status, consistent across health states and geographic locations. We classified 172 gut microbial species as co-colonizers and 135 as co-excluders, revealing a genus-wide signal of colonization resistance within Faecalibacterium and strain-specific co-colonization patterns of the underexplored Faecalimonas phoceensis. Co-exclusion is linked to functions involved in short-chain fatty acid production, iron metabolism and quorum sensing, while co-colonization is linked to greater functional diversity and metabolic resemblance to Enterobacteriaceae. Our work underscores the critical role of the intestinal environment in the colonization success of gut-associated opportunistic pathogens with implications for developing non-antibiotic therapeutic strategies.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interests.
Figures













References
-
- Leshem, A., Liwinski, T. & Elinav, E. Immune–microbiota interplay and colonization resistance in infection. Mol. Cell78, 597–613 (2020). - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
