

Genomic characterization of Huntington's disease genetic modifiers informs drug target tractability
- PMID: 39801710
- PMCID: PMC11724427
- DOI: 10.1093/braincomms/fcae418
Genomic characterization of Huntington's disease genetic modifiers informs drug target tractability
Abstract
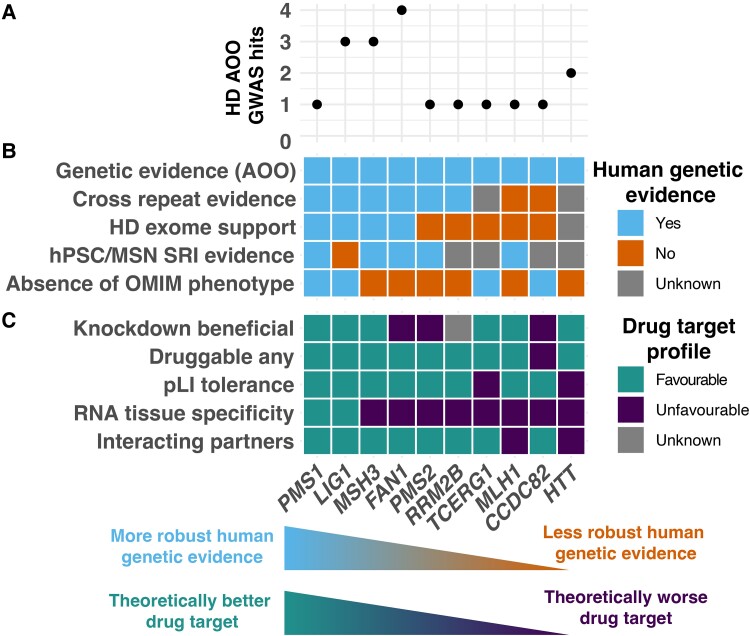
Huntington's disease is caused by a CAG repeat in the HTT gene. Repeat length correlates inversely with the age of onset but only explains part of the observed clinical variability. Genome-wide association studies highlight DNA repair genes in modifying disease onset, but further research is required to identify causal genes and evaluate their tractability as drug targets. To address these gaps and learn important preclinical information, we analysed genome-wide association study data from a large Huntington's disease age-of-onset study (n = 9064), prioritizing robust candidate Huntington's disease modifier genes using bioinformatic approaches and analysing related information for these genes from large-scale human genetic repositories. We supplemented this information with other Huntington's disease-related screens, including exome studies of Huntington's disease onset and high-throughput assessments of mHTT toxicity. To confirm whether Huntington's disease modifiers are shared across repeat expansion disorders, we also analysed age-of-onset genome-wide association study data from X-linked dystonia-parkinsonism caused by a (CCCTCT)n expansion. We also studied modifier-related associations with rare diseases to inform potential off-target therapeutic effects and conducted comprehensive phenome-wide studies to identify other traits linked to these genes. Finally, we evaluated the aggregated human genetic evidence and theoretical druggability of the prioritized Huntington's disease modifier genes, including characteristics recently associated with clinical trial stoppage due to safety concerns (i.e. human genetic constraint, number of interacting partners and RNA tissue expression specificity). In total, we annotated and assessed nine robust candidate Huntington's disease modifier genes. Notably, we detected a high correlation (R 2 = 0.78) in top age-of-onset genome-wide association study hits across repeat expansion disorders, emphasizing cross-disorder relevance. Clinical genetic repositories analysis showed DNA repair genes, such as MLH1, PMS2 and MSH3, are associated with cancer phenotypes, suggesting potential limitations as drug targets. LIG1 and RRM2B were both associated with neurofibrillary tangles, which may provide a link to a potential role in mHTT aggregates, while MSH3 was associated with several cortical morphology-related traits relevant to Huntington's disease. Finally, human genetic evidence and theoretical druggability analyses prioritized and ranked modifier genes, with PMS1 exhibiting the most favourable profile. Notably, HTT itself ranked poorly as a theoretical drug target, emphasizing the importance of exploring modifier-based alternative targets. In conclusion, our study highlights the importance of human genomic information to prioritize Huntington's disease modifier genes as drug targets, providing a basis for future therapeutic development in Huntington's disease and other repeat expansion disorders.
Keywords: human genetics; monogenic disease; repeat expansion.
© The Author(s) 2025. Published by Oxford University Press on behalf of the Guarantors of Brain.
Conflict of interest statement
The authors report no competing interests.
Figures




Similar articles
-
Interrupting sequence variants and age of onset in Huntington's disease: clinical implications and emerging therapies.Lancet Neurol. 2020 Nov;19(11):930-939. doi: 10.1016/S1474-4422(20)30343-4. Lancet Neurol. 2020. PMID: 33098802 Review.
-
In vivo CRISPR-Cas9 genome editing in mice identifies genetic modifiers of somatic CAG repeat instability in Huntington's disease.Nat Genet. 2025 Feb;57(2):314-322. doi: 10.1038/s41588-024-02054-5. Epub 2025 Jan 22. Nat Genet. 2025. PMID: 39843658 Free PMC article.
-
Modification of Huntington's disease by short tandem repeats.Brain Commun. 2024 Jan 23;6(2):fcae016. doi: 10.1093/braincomms/fcae016. eCollection 2024. Brain Commun. 2024. PMID: 38449714 Free PMC article.
-
Promotion of somatic CAG repeat expansion by Fan1 knock-out in Huntington's disease knock-in mice is blocked by Mlh1 knock-out.Hum Mol Genet. 2020 Nov 4;29(18):3044-3053. doi: 10.1093/hmg/ddaa196. Hum Mol Genet. 2020. PMID: 32876667 Free PMC article.
-
Modifiers of Somatic Repeat Instability in Mouse Models of Friedreich Ataxia and the Fragile X-Related Disorders: Implications for the Mechanism of Somatic Expansion in Huntington's Disease.J Huntingtons Dis. 2021;10(1):149-163. doi: 10.3233/JHD-200423. J Huntingtons Dis. 2021. PMID: 33579860 Free PMC article. Review.
References
-
- The Huntington’s Disease Collaborative Research Group . A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72(6):971–983. - PubMed
-
- Bates GP, Dorsey R, Gusella JF, et al. . Huntington disease. Nat Rev Dis Primer. 2015;1:15005. - PubMed
-
- Wright GEB, Findlay Black H, Collins JA, et al. . Interrupting sequence variants and age of onset in Huntington’s disease: Clinical implications and emerging therapies. Lancet Neurol. 2020;19(11):930–939. - PubMed
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous