

De novo generation of dual-target compounds using artificial intelligence
- PMID: 39801837
- PMCID: PMC11721219
- DOI: 10.1016/j.isci.2024.111526
De novo generation of dual-target compounds using artificial intelligence
Abstract
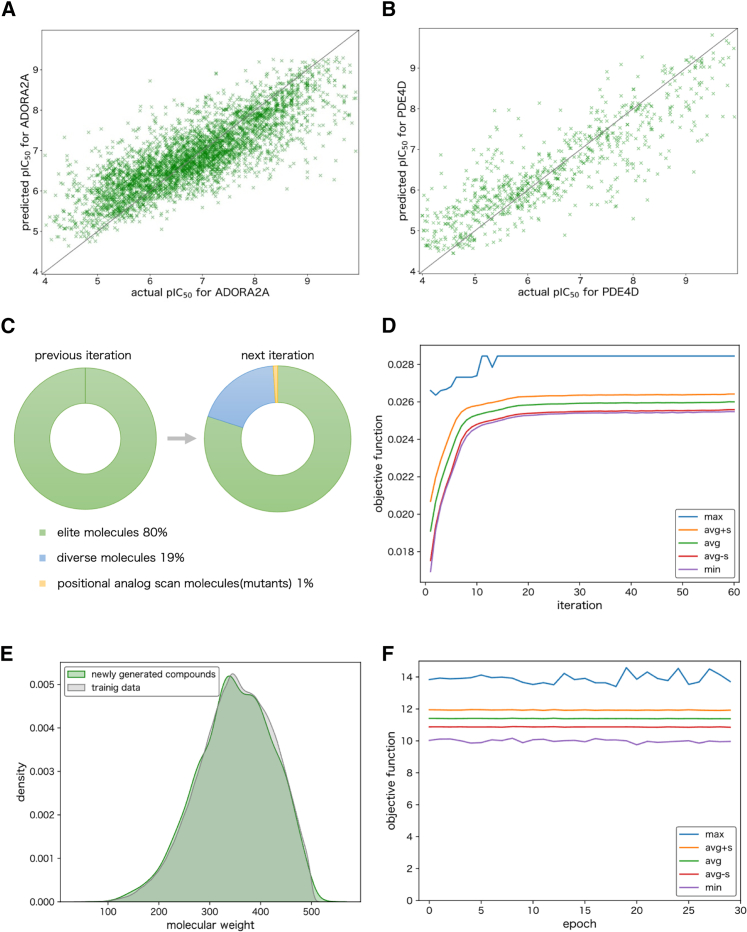
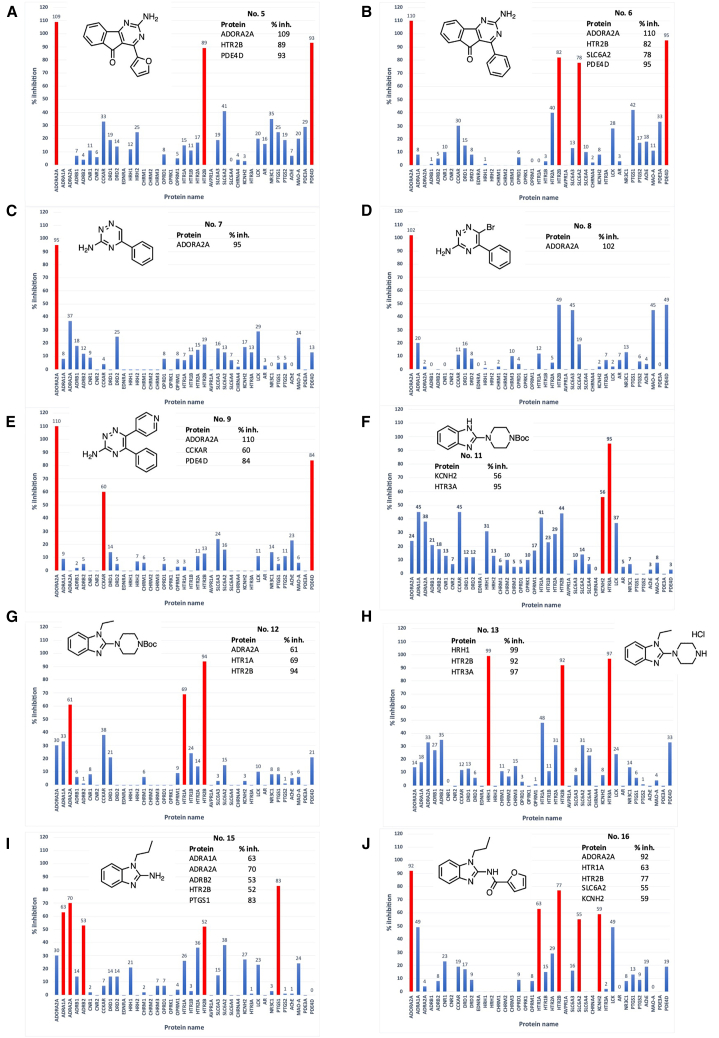
Drugs that interact with multiple therapeutic targets are potential high-value products in polypharmacology-based drug discovery, but the rational design remains a formidable challenge. Here, we present artificial intelligence (AI)-based methods to design the chemical structures of compounds that interact with multiple therapeutic target proteins. The molecular structure generation is performed by a fragment-based approach using a genetic algorithm with chemical substructures and a deep learning approach using reinforcement learning with stochastic policy gradients in the framework of generative adversarial networks. Using the proposed methods, we designed the chemical structures of compounds that would interact with two therapeutic targets of bronchial asthma, i.e., adenosine A2a receptor (ADORA2A) and phosphodiesterase 4D (PDE4D). We then synthesized 10 compounds and evaluated their bioactivities via the binding assays of 39 target human proteins, including ADORA2A and PDE4D. Three of the 10 synthesized compounds successfully interacted with ADORA2A and PDE4D with high specificity.
Keywords: Bioinformatics; Biological sciences; Natural sciences; Pharmacoinformatics.
© 2024 The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures




Similar articles
-
De novo generation of dual-target ligands using adversarial training and reinforcement learning.Brief Bioinform. 2021 Nov 5;22(6):bbab333. doi: 10.1093/bib/bbab333. Brief Bioinform. 2021. PMID: 34410338
-
Advanced AI and ML frameworks for transforming drug discovery and optimization: With innovative insights in polypharmacology, drug repurposing, combination therapy and nanomedicine.Eur J Med Chem. 2025 Feb 15;284:117164. doi: 10.1016/j.ejmech.2024.117164. Epub 2024 Dec 13. Eur J Med Chem. 2025. PMID: 39721292 Review.
-
Artificial intelligence to deep learning: machine intelligence approach for drug discovery.Mol Divers. 2021 Aug;25(3):1315-1360. doi: 10.1007/s11030-021-10217-3. Epub 2021 Apr 12. Mol Divers. 2021. PMID: 33844136 Free PMC article. Review.
-
De novo generation of multi-target compounds using deep generative chemistry.Nat Commun. 2024 May 6;15(1):3636. doi: 10.1038/s41467-024-47120-y. Nat Commun. 2024. PMID: 38710699 Free PMC article.
-
De novo generation of dual-target ligands for the treatment of SARS-CoV-2 using deep learning, virtual screening, and molecular dynamic simulations.J Biomol Struct Dyn. 2024 Apr;42(6):3019-3029. doi: 10.1080/07391102.2023.2234481. Epub 2023 Jul 14. J Biomol Struct Dyn. 2024. PMID: 37449757
Cited by
-
Phenotypic Drug Discovery Platform by Quantitative High-Throughput Screening Identifies Antiapoptotic Molecules in a Zebrafish Model of Age-Related Macular Degeneration.ACS Omega. 2025 Jul 7;10(28):30467-30488. doi: 10.1021/acsomega.5c02227. eCollection 2025 Jul 22. ACS Omega. 2025. PMID: 40727801 Free PMC article.
References
-
- Kusner M.J., Paige B., Hernández-Lobato J.M. Vol. 70. 2017. Grammar variational autoencoder; pp. 1945–1954. (Proceedings of the 34th International Conference on Machine Learning). - DOI
-
- Dai H., Tian Y., Dai B., Skiena S., Song L. Proceedings of the International Conference on Learning Representations. 2018. Syntax-directed variational autoencoder for molecule generation. - DOI
LinkOut - more resources
Full Text Sources
