

This is a preprint.
Mapping chromatin interactions at melanoma susceptibility loci and cell-type specific dataset integration uncovers distant gene targets of cis-regulation
- PMID: 39802764
- PMCID: PMC11722502
- DOI: 10.1101/2024.11.14.24317204
Mapping chromatin interactions at melanoma susceptibility loci and cell-type specific dataset integration uncovers distant gene targets of cis-regulation
Update in
-
Mapping chromatin interactions at melanoma susceptibility loci uncovers distant cis-regulatory gene targets.Am J Hum Genet. 2025 Jul 3;112(7):1625-1648. doi: 10.1016/j.ajhg.2025.04.015. Epub 2025 May 22. Am J Hum Genet. 2025. PMID: 40409268 Free PMC article.
Abstract
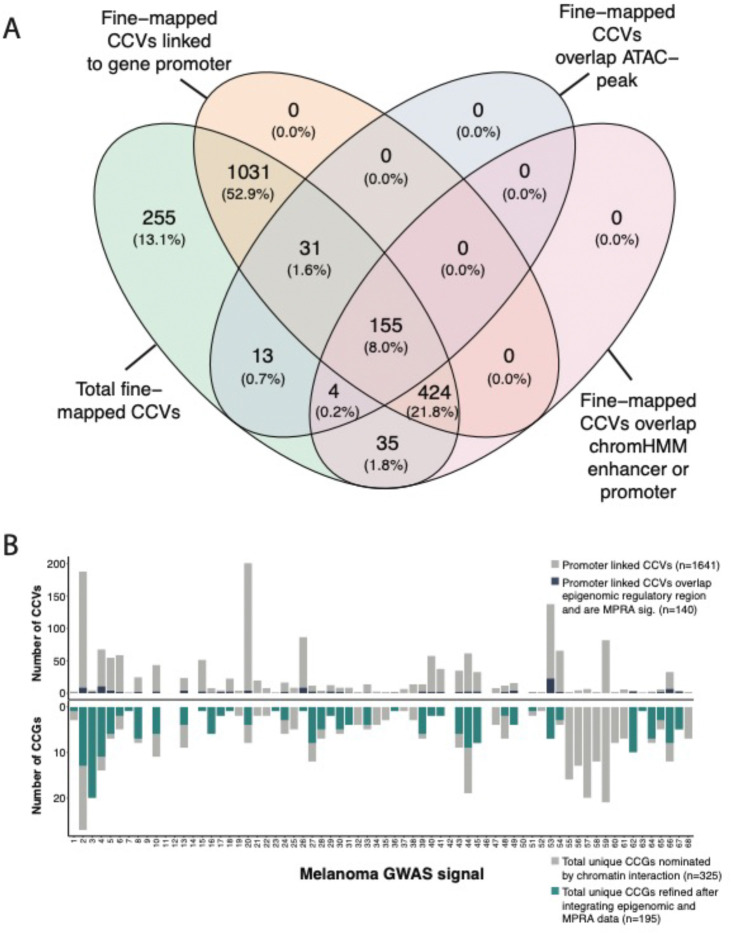
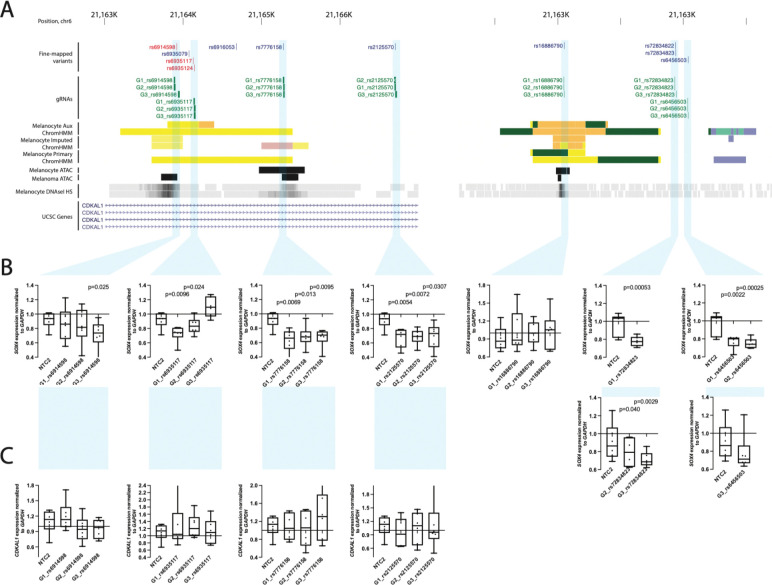
Genome-wide association studies (GWAS) of melanoma risk have identified 68 independent signals at 54 loci. For most loci, specific functional variants and their respective target genes remain to be established. Capture-HiC is an assay that links fine-mapped risk variants to candidate target genes by comprehensively mapping cell-type specific chromatin interactions. We performed a melanoma GWAS region-focused capture-HiC assay in human primary melanocytes to identify physical interactions between fine-mapped risk variants and potential causal melanoma susceptibility genes. Overall, chromatin interaction data alone nominated potential causal genes for 61 of the 68 melanoma risk signals, identifying many candidates beyond those reported by previous studies. We further integrated these data with cell-type specific epigenomic (chromatin state, accessibility), gene expression (eQTL/TWAS), DNA methylation (meQTL/MWAS), and massively parallel reporter assay (MPRA) data to prioritize potentially cis-regulatory variants and their respective candidate gene targets. From the set of fine-mapped variants across these loci, we identified 140 prioritized candidate causal variants linked to 195 candidate genes at 42 risk signals. In addition, we developed an integrative scoring system to facilitate candidate gene prioritization, integrating melanocyte and melanoma datasets. Notably, at several GWAS risk signals we observed long-range chromatin connections (500 kb to >1 Mb) with distant candidate target genes. We validated several such cis-regulatory interactions using CRISPR inhibition, providing evidence for known cancer driver genes MDM4 and CBL, as well as the SRY-box transcription factor SOX4, as likely melanoma risk genes.
Conflict of interest statement
DECLARATION OF INTERESTS The authors declare no competing interests.
Figures





References
Publication types
LinkOut - more resources
Full Text Sources
Miscellaneous