

This is a preprint.
CEBPA repression by MECOM blocks differentiation to drive aggressive leukemias
- PMID: 39803492
- PMCID: PMC11722404
- DOI: 10.1101/2024.12.30.630680
CEBPA repression by MECOM blocks differentiation to drive aggressive leukemias
Update in
-
CEBPA repression by MECOM blocks differentiation to drive aggressive leukemias.Blood. 2025 Dec 18;146(25):3019-3035. doi: 10.1182/blood.2025028954. Blood. 2025. PMID: 40991835 Free PMC article.
Abstract
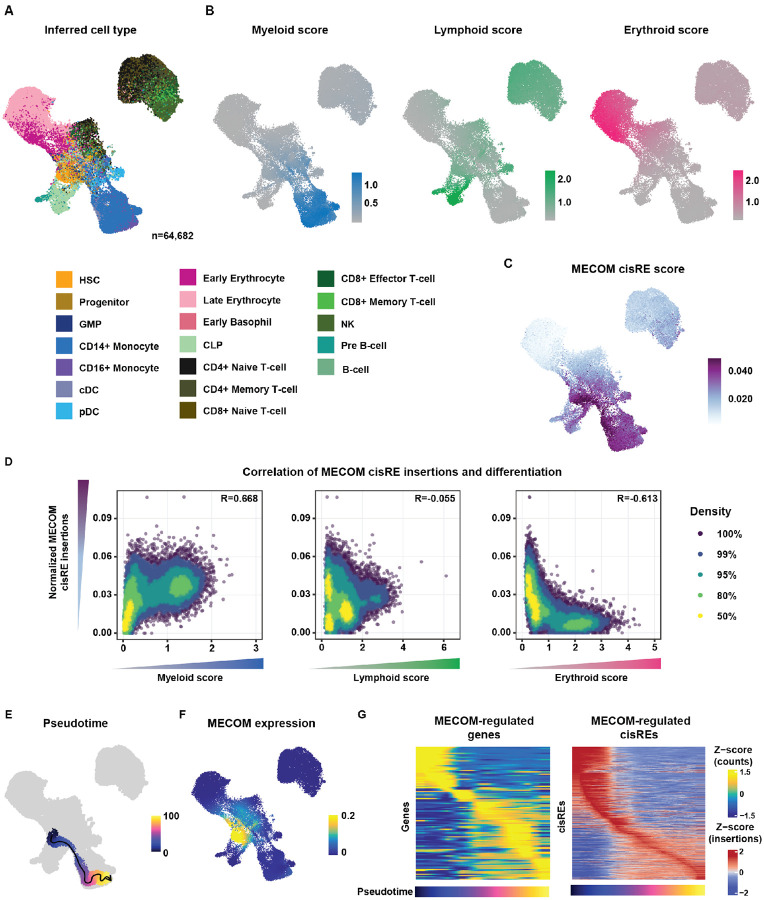
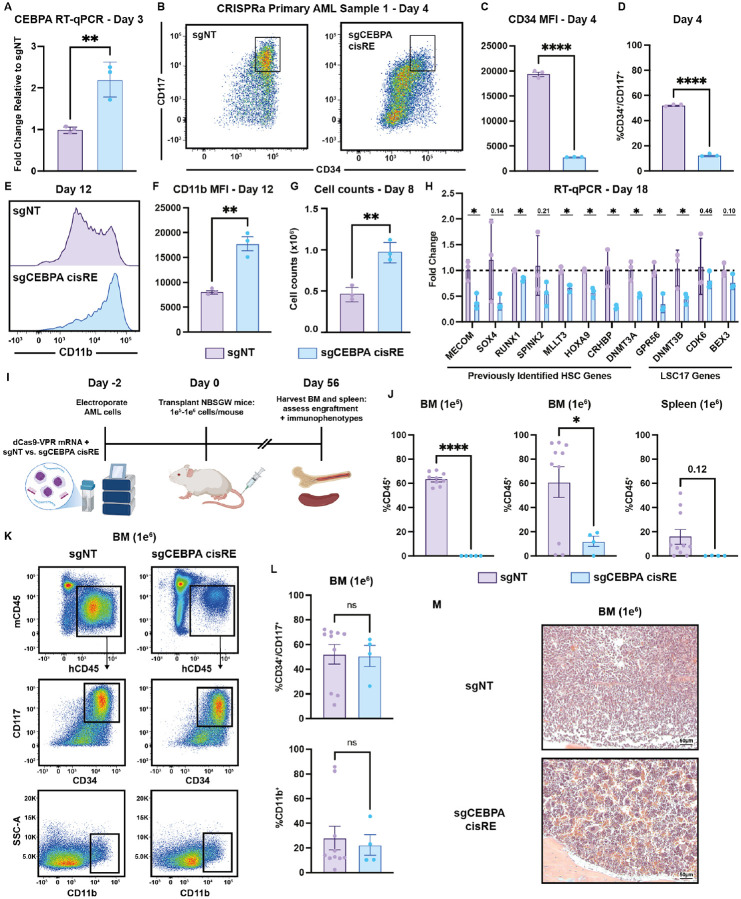
Acute myeloid leukemias (AMLs) have an overall poor prognosis with many high-risk cases co-opting stem cell gene regulatory programs, yet the mechanisms through which this occurs remain poorly understood. Increased expression of the stem cell transcription factor, MECOM, underlies one key driver mechanism in largely incurable AMLs. How MECOM results in such aggressive AML phenotypes remains unknown. To address existing experimental limitations, we engineered and applied targeted protein degradation with functional genomic readouts to demonstrate that MECOM promotes malignant stem cell-like states by directly repressing pro-differentiation gene regulatory programs. Remarkably and unexpectedly, a single node in this network, a MECOM-bound cis-regulatory element located 42 kb downstream of the myeloid differentiation regulator CEBPA, is both necessary and sufficient for maintaining MECOM-driven leukemias. Importantly, targeted activation of this regulatory element promotes differentiation of these aggressive AMLs and reduces leukemia burden in vivo, suggesting a broadly applicable differentiation-based approach for improving therapy.
Figures





References
-
- Döhner H., Weisdorf D. J. & Bloomfield C. D. Acute myeloid leukemia. N. Engl. J. Med. 373, 1136–1152 (2015). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources