

This is a preprint.
Organ injury accelerates stem cell differentiation by modulating a fate-transducing lateral inhibition circuit
- PMID: 39803552
- PMCID: PMC11722240
- DOI: 10.1101/2024.12.29.630675
Organ injury accelerates stem cell differentiation by modulating a fate-transducing lateral inhibition circuit
Abstract
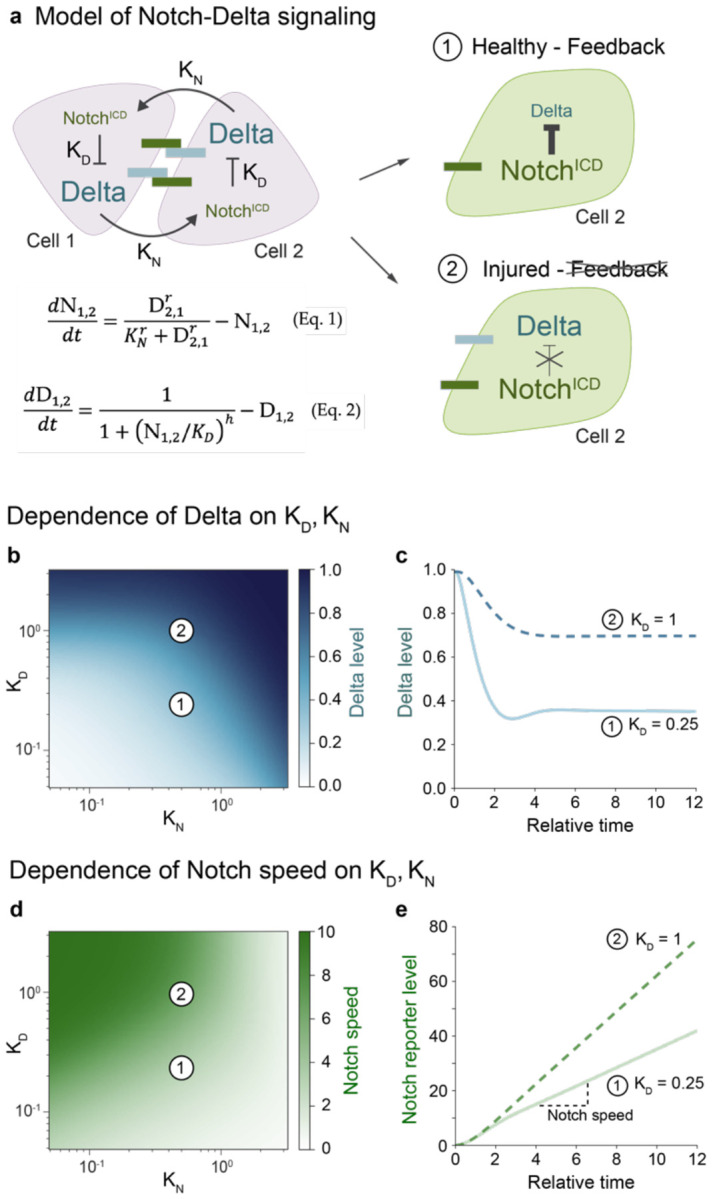
To rebuild tissue form and function, injured organs accelerate the differentiation of replacement stem cell progeny. Here we demonstrate that injury-induced factors open the throttle on faster differentiation by streamlining the archetypal signaling circuit that patterns cell fates. During normal turnover of the adult Drosophila intestine, fates are patterned by a conserved lateral inhibition circuit: In stem cell pairs, mutual activation of Notch receptor by Delta ligand feeds back to create opposing states of high Notch/low Delta and low Notch/high Delta; cells terminally differentiate once their Notch activity exceeds a fate-deciding threshold. After feeding flies a gut-damaging toxin, we perform in vivo imaging of real-time intestinal repair and trace Notch reporter dynamics in single cells. We find that tissue damage causes the speed of Notch signal activation to accelerate dramatically; faster activation expedites terminal differentiation by propelling cells past the critical Notch threshold more quickly. Combining single-cell analyses with dynamical modeling, we show that faster activation results from aberrant elevation of Delta ligand due to loss of time-delaying circuit feedback. Injury abolishes feedback via a cytokine-JAK-STAT relay from damaged cells to stem cells, causing stem cells to deactivate the Notch co-repressor that normally turns off Delta. Thus, organ injury unmasks latent plasticity in Notch-Delta lateral inhibition to propel the differentiation of new replacement cells. By unifying temporal and spatial fate control in a single, adaptable signaling circuit, organs tune stem cell dynamics to meet environmental challenges.
Figures






References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous