

Robust collection and processing for label-free single voxel proteomics
- PMID: 39805815
- PMCID: PMC11730317
- DOI: 10.1038/s41467-024-54643-x
Robust collection and processing for label-free single voxel proteomics
Abstract
With advanced mass spectrometry (MS)-based proteomics, genome-scale proteome coverage can be achieved from bulk tissues. However, such bulk measurement lacks spatial resolution and obscures tissue heterogeneity, precluding proteome mapping of tissue microenvironment. Here we report an integrated wet collection of single microscale tissue voxels and Surfactant-assisted One-Pot voxel processing method termed wcSOP for robust label-free single voxel proteomics. wcSOP capitalizes on buffer droplet-assisted wet collection of single voxels dissected by LCM to the tube cap and SOP voxel processing in the same collection cap. This method enables reproducible, label-free quantification of approximately 900 and 4600 proteins for single voxels at 20 µm × 20 µm × 10 µm (~1 cell region) and 200 µm × 200 µm × 10 µm (~100 cell region) from fresh frozen human spleen tissue, respectively. It can reveal spatially resolved protein signatures and region-specific signaling pathways. Furthermore, wcSOP-MS is demonstrated to be broadly applicable for OCT-embedded and FFPE human archived tissues as well as for small-scale 2D proteome mapping of tissues at high spatial resolutions. wcSOP-MS may pave the way for routine robust single voxel proteomics and spatial proteomics.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: The authors declare no competing interest.
Figures
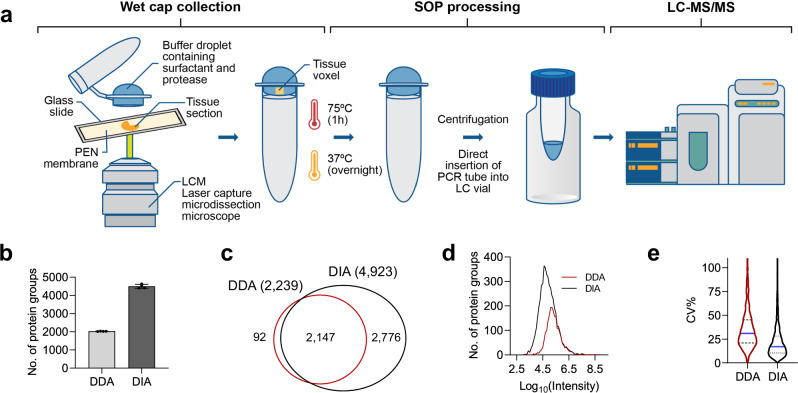


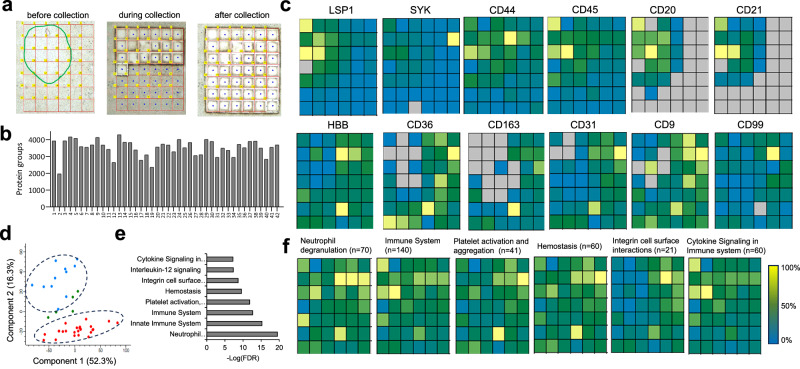

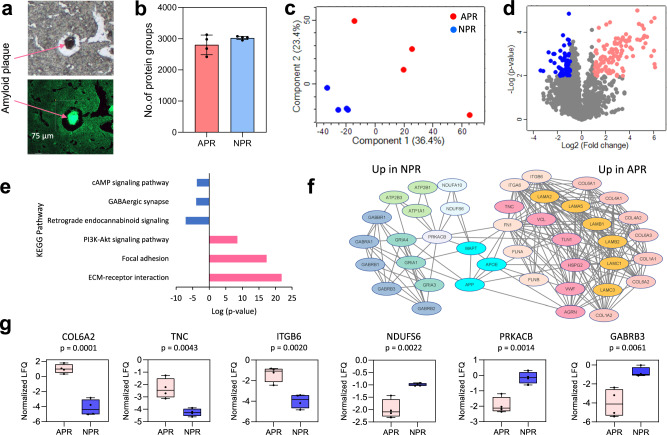
Update of
-
Robust collection and processing for label-free single voxel proteomics.bioRxiv [Preprint]. 2023 Aug 15:2023.08.14.553333. doi: 10.1101/2023.08.14.553333. bioRxiv. 2023. Update in: Nat Commun. 2025 Jan 13;16(1):547. doi: 10.1038/s41467-024-54643-x. PMID: 37645907 Free PMC article. Updated. Preprint.
References
MeSH terms
Substances
Grants and funding
- P41 GM103493/GM/NIGMS NIH HHS/United States
- RF1MH128885/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- R01 CA245699/CA/NCI NIH HHS/United States
- R01 GM139858/GM/NIGMS NIH HHS/United States
- UH3CA256967/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
LinkOut - more resources
Full Text Sources
Miscellaneous
