

Loss of Kmt2c or Kmt2d primes urothelium for tumorigenesis and redistributes KMT2A-menin to bivalent promoters
- PMID: 39806204
- PMCID: PMC11735410
- DOI: 10.1038/s41588-024-02015-y
Loss of Kmt2c or Kmt2d primes urothelium for tumorigenesis and redistributes KMT2A-menin to bivalent promoters
Abstract
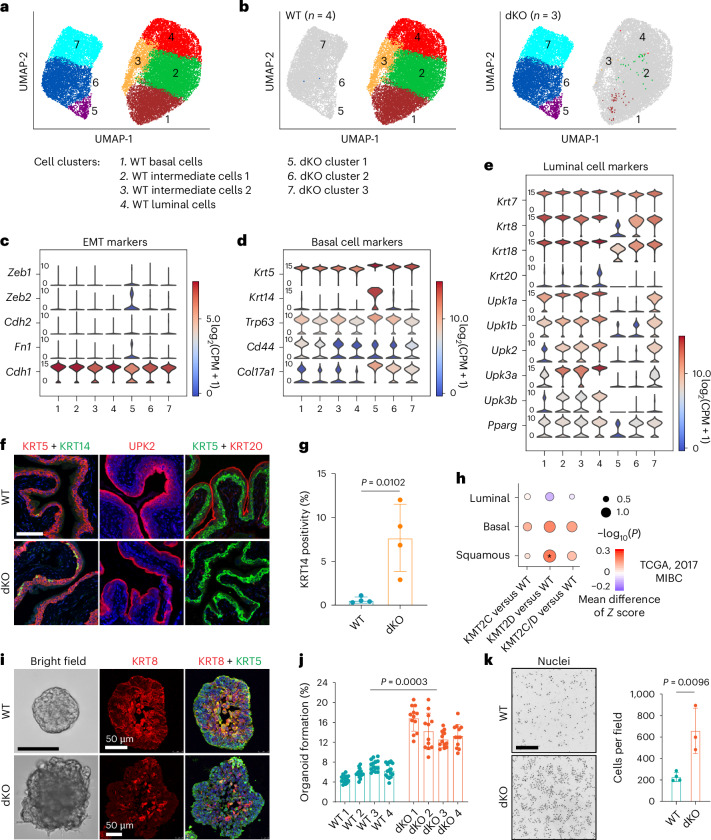
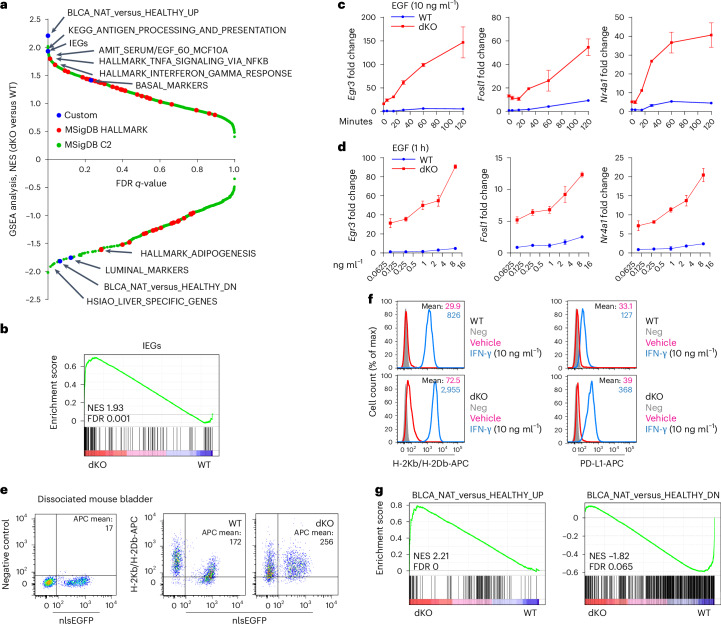
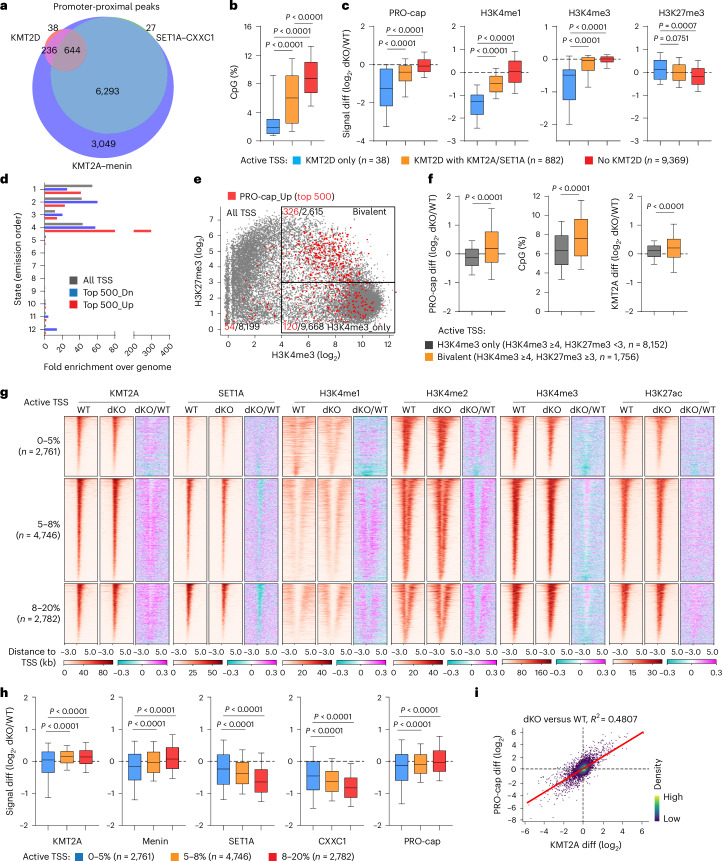
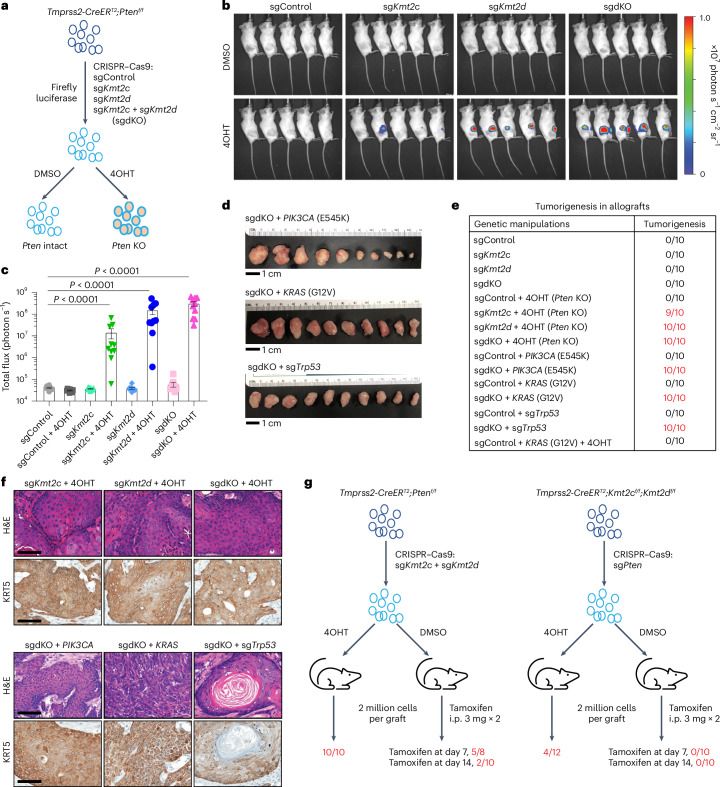
Members of the KMT2C/D-KDM6A complex are recurrently mutated in urothelial carcinoma and in histologically normal urothelium. Here, using genetically engineered mouse models, we demonstrate that Kmt2c/d knockout in the urothelium led to impaired differentiation, augmented responses to growth and inflammatory stimuli and sensitization to oncogenic transformation by carcinogen and oncogenes. Mechanistically, KMT2D localized to active enhancers and CpG-poor promoters that preferentially regulate the urothelial lineage program and Kmt2c/d knockout led to diminished H3K4me1, H3K27ac and nascent RNA transcription at these sites, which leads to impaired differentiation. Kmt2c/d knockout further led to KMT2A-menin redistribution from KMT2D localized enhancers to CpG-high and bivalent promoters, resulting in derepression of signal-induced immediate early genes. Therapeutically, Kmt2c/d knockout upregulated epidermal growth factor receptor signaling and conferred vulnerability to epidermal growth factor receptor inhibitors. Together, our data posit that functional loss of Kmt2c/d licenses a molecular 'field effect' priming histologically normal urothelium for oncogenic transformation and presents therapeutic vulnerabilities.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: P.C. has received personal honoraria/advisory boards/consulting fees from Deciphera, Exelixis, Zai Lab, Novartis and Ningbo NewBay Medical Technology. P.C. has received institutional research funding from Pfizer/Array, Novartis, Deciphera and Ningbo NewBay Medical Technology. Y.C. has stock ownership and received royalties from Oric Pharmaceuticals. S.C. has received institutional research support from Daiichi-Sankyo, Paige.ai and AstraZeneca, is founder/shareholder of Odyssey Biosciences, has shares in Totus Medicines, and personal consulting fees from Sanofi, Novartis, Inivata, Lilly and AstraZeneca. J.E.R. has received personal honoraria/consulting fees from BMS, Merck, Pfizer, Pharmacyclics, Boheringer Ingelheim, EMD-Serono, GSK, Infinity, Janssen, Mirati, BioClin, Lilly, Tyra Biosciences, Astellas, Seagen, Bayer, AstraZeneca, Roche/Genentech and QED Therapeutics, and conducted trials from Astellas, Seagen, Bayer, AstraZeneca, Roche/Genentech and QED Therapeutics. S.R.S. is an equity holder and member of the scientific advisory board of NeuScience, Inc. and a consultant at Third Bridge Group Limited. D.B.S. has consulted/received honoraria from Rain, Pfizer, Fog Pharma, PaigeAI, BridgeBio, Scorpion Therapeutics, FORE Therapeutics, Function Oncology, Pyramid and Elsie Biotechnologies, Inc. None of these disclosures are directly related to this study. The other authors declare no competing interests.
Figures














References
-
- Sanli, O. et al. Bladder cancer. Nat. Rev. Dis. Primers3, 17022 (2017). - PubMed
-
- Simon, R. et al. Cytogenetic analysis of multifocal bladder cancer supports a monoclonal origin and intraepithelial spread of tumor cells. Cancer Res.61, 355–362 (2001). - PubMed
-
- Ramal, M., Corral, S., Kalisz, M., Lapi, E. & Real, F. X. The urothelial gene regulatory network: understanding biology to improve bladder cancer management. Oncogene43, 1–21 (2024). - PubMed
MeSH terms
Substances
Grants and funding
- R01CA193837/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- R01 CA193837/CA/NCI NIH HHS/United States
- W81XWH-15-1-0124/U.S. Department of Defense (United States Department of Defense)
- P50 CA092629/CA/NCI NIH HHS/United States
- R01 CA233899/CA/NCI NIH HHS/United States
- P30 CA008748/CA/NCI NIH HHS/United States
- P50 CA221745/CA/NCI NIH HHS/United States
- DP2CA174499/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- R01 CA208100/CA/NCI NIH HHS/United States
- P50 CA217694/CA/NCI NIH HHS/United States
- R01CA208100/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- U54 CA224079/CA/NCI NIH HHS/United States
- DP2 CA174499/CA/NCI NIH HHS/United States
- R01CA228216/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- P01 CA221757/CA/NCI NIH HHS/United States
- R01 CA265026/CA/NCI NIH HHS/United States
- R01CA233899/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- R01 CA228216/CA/NCI NIH HHS/United States
- P50CA221745/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- U01CA224044/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- R01 CA280657/CA/NCI NIH HHS/United States
- P01CA221757/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- P50CA217694/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- P50CA092629/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- U01 CA224044/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
