

Harnessing the tumor microenvironment: targeted cancer therapies through modulation of epithelial-mesenchymal transition
- PMID: 39806516
- PMCID: PMC11733683
- DOI: 10.1186/s13045-024-01634-6
Harnessing the tumor microenvironment: targeted cancer therapies through modulation of epithelial-mesenchymal transition
Abstract
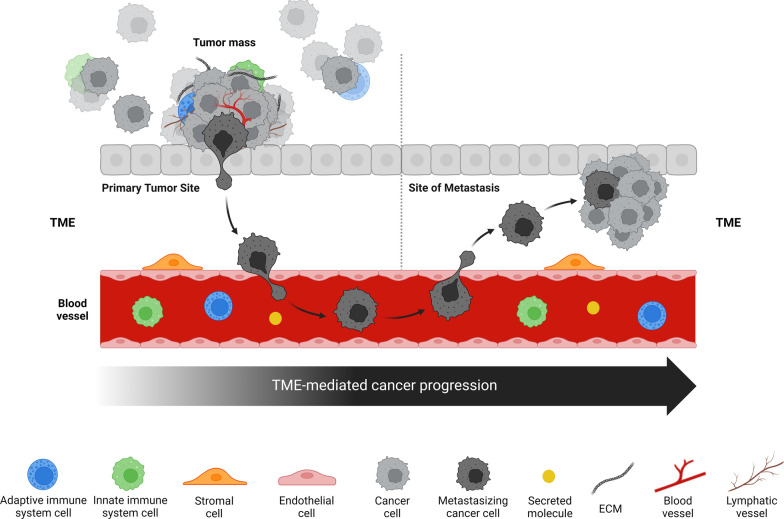
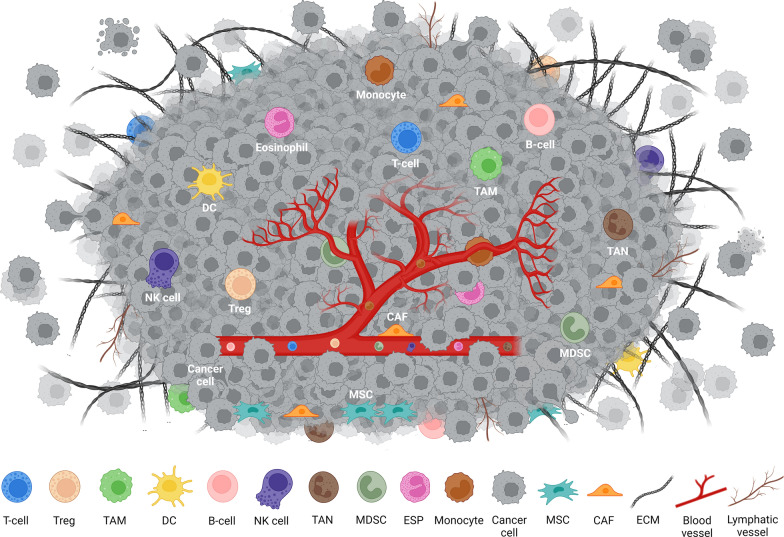
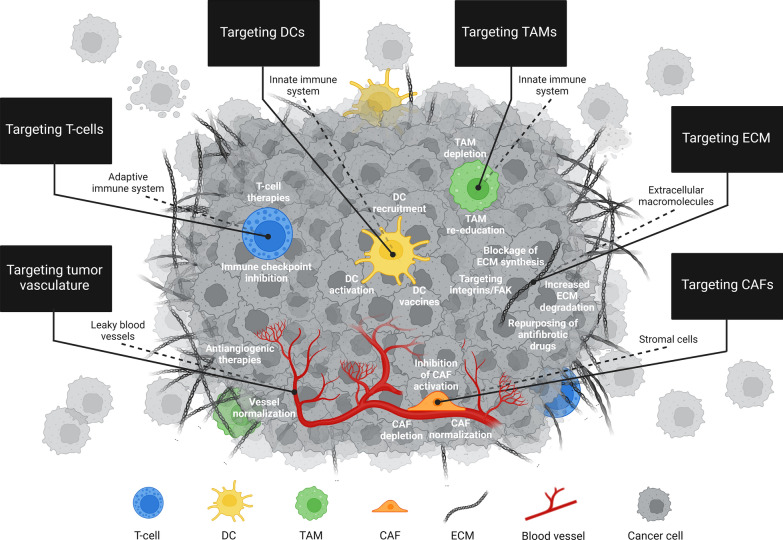
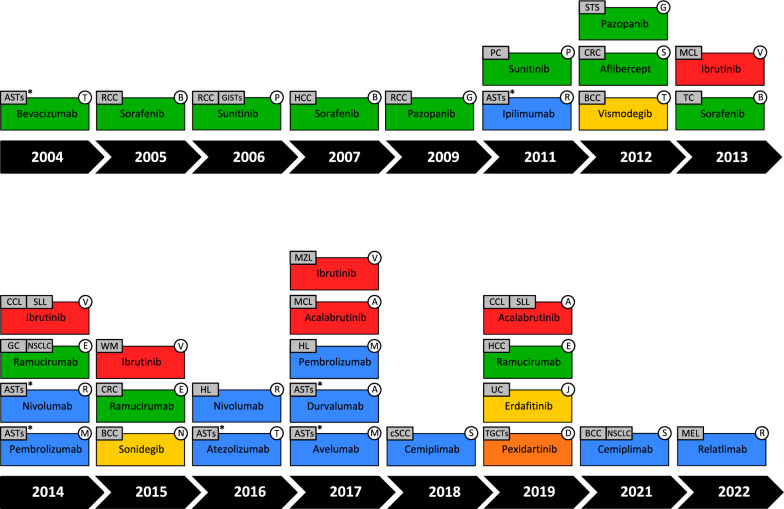
The tumor microenvironment (TME) is integral to cancer progression, impacting metastasis and treatment response. It consists of diverse cell types, extracellular matrix components, and signaling molecules that interact to promote tumor growth and therapeutic resistance. Elucidating the intricate interactions between cancer cells and the TME is crucial in understanding cancer progression and therapeutic challenges. A critical process induced by TME signaling is the epithelial-mesenchymal transition (EMT), wherein epithelial cells acquire mesenchymal traits, which enhance their motility and invasiveness and promote metastasis and cancer progression. By targeting various components of the TME, novel investigational strategies aim to disrupt the TME's contribution to the EMT, thereby improving treatment efficacy, addressing therapeutic resistance, and offering a nuanced approach to cancer therapy. This review scrutinizes the key players in the TME and the TME's contribution to the EMT, emphasizing avenues to therapeutically disrupt the interactions between the various TME components. Moreover, the article discusses the TME's implications for resistance mechanisms and highlights the current therapeutic strategies toward TME modulation along with potential caveats.
Keywords: Cancer; Cancer-associated fibroblasts (CAFs); Chimeric antigen-receptor (CAR) T-cell therapy; Dendritic cells (DCs); Epithelial-mesenchymal transition (EMT); Extracellular matrix (ECM); Metastasis; Myeloid-derived suppressor cells (MDSCs); Natural killer (NK) cells; T-cell receptor (TCR) therapy; T-cells, B-cells, tumor-associated macrophages (TAMs); Theranostics; Tumor microenvironment (TME); Tumor-associated neutrophils (TANs).
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethics approval and consent to participate: Not Applicable. Consent for publication: We confirm that the manuscript has been read and approved by all named authors and that there are no other persons who satisfied the criteria for authorship but are not listed. We further confirm that the order of authors listed in the manuscript has been approved by all the authors. All figures are original and were drawn using BioRender by the authors for this review. Competing interests: The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work in this paper. M.H.A. is founder, shareholder and scientific advisor of IO Biotech ApS. LMC is a consultant for Evergreen Theragnostics. S.K. has consulted for Telix Pharmaceuticals Ltd., acknowledges support for investigator services from RayzeBio and holds the following patent: PCT/US2021/039418 (THOR cell (tumor homing radio-emitting cell).
Figures




References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
