

Revealing the reaction path of UVC bond rupture in cyclic disulfides with ultrafast x-ray scattering
- PMID: 39813348
- PMCID: PMC11734709
- DOI: 10.1126/sciadv.adp9175
Revealing the reaction path of UVC bond rupture in cyclic disulfides with ultrafast x-ray scattering
Abstract
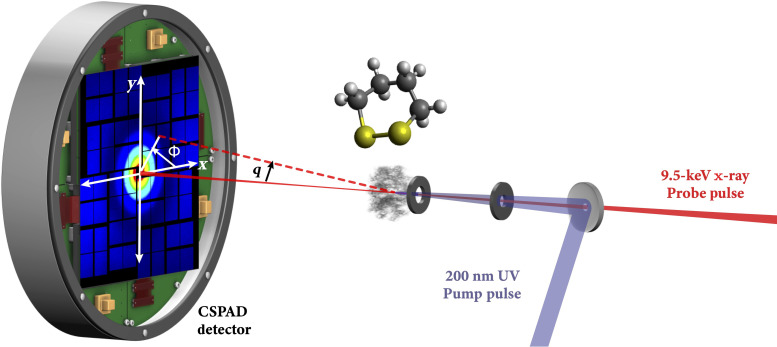
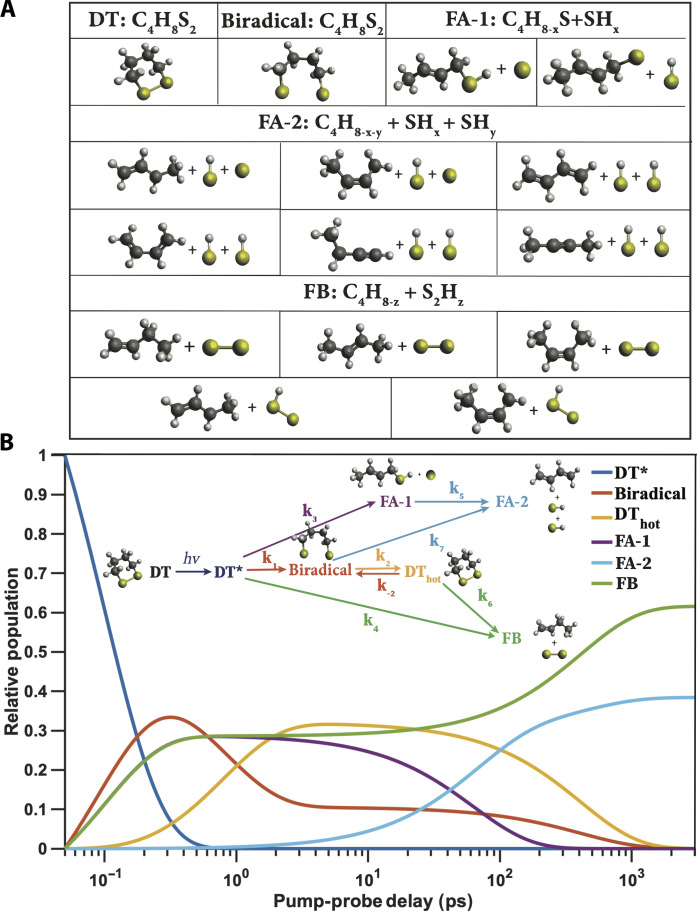
Disulfide bonds are ubiquitous molecular motifs that influence the tertiary structure and biological functions of many proteins. Yet, it is well known that the disulfide bond is photolabile when exposed to ultraviolet C (UVC) radiation. The deep-UV-induced S─S bond fragmentation kinetics on very fast timescales are especially pivotal to fully understand the photostability and photodamage repair mechanisms in proteins. In 1,2-dithiane, the smallest saturated cyclic molecule that mimics biologically active species with S─S bonds, we investigate the photochemistry upon 200-nm excitation by femtosecond time-resolved x-ray scattering in the gas phase using an x-ray free electron laser. In the femtosecond time domain, we find a very fast reaction that generates molecular fragments with one and two sulfur atoms. On picosecond and nanosecond timescales, a complex network of reactions unfolds that, ultimately, completes the sulfur dissociation from the parent molecule.
Figures



References
-
- Creighton T. E., Disulphide bonds and protein stability. BioEssays 8, 57–63 (1988). - PubMed
-
- Sevier C. S., Kaiser C. A., Formation and transfer of disulphide bonds in living cells. Nat. Rev. Mol. Cell Biol. 3, 836–847 (2002). - PubMed
-
- Crespo-Hernández C. E., Cohen B., Kohler B., Base stacking controls excited-state dynamics in A· T DNA. Nature 436, 1141–1144 (2005). - PubMed
