

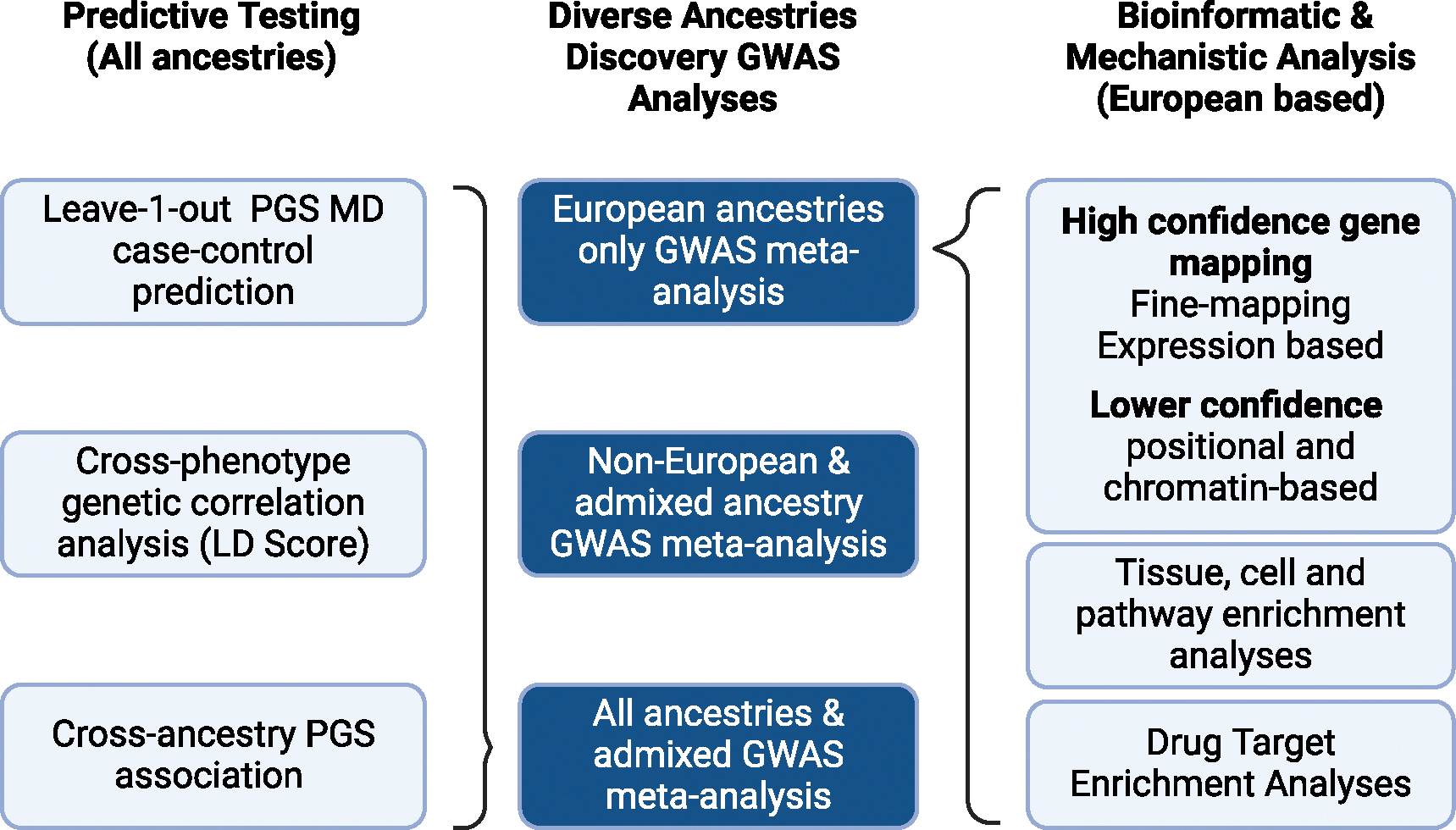
Trans-ancestry genome-wide study of depression identifies 697 associations implicating cell types and pharmacotherapies
- PMID: 39814019
- PMCID: PMC11829167
- DOI: 10.1016/j.cell.2024.12.002
Trans-ancestry genome-wide study of depression identifies 697 associations implicating cell types and pharmacotherapies
Abstract
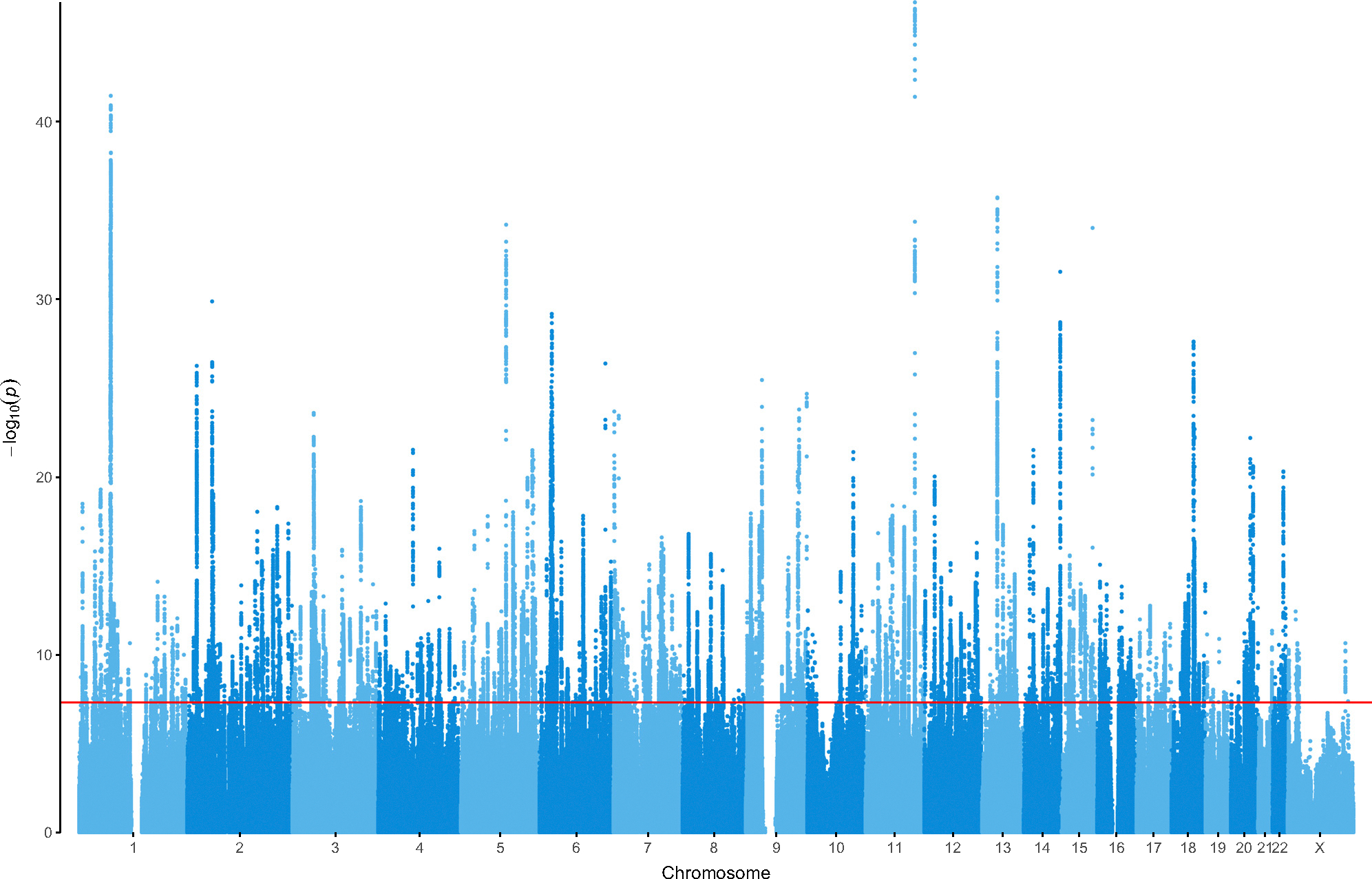
In a genome-wide association study (GWAS) meta-analysis of 688,808 individuals with major depression (MD) and 4,364,225 controls from 29 countries across diverse and admixed ancestries, we identify 697 associations at 635 loci, 293 of which are novel. Using fine-mapping and functional tools, we find 308 high-confidence gene associations and enrichment of postsynaptic density and receptor clustering. A neural cell-type enrichment analysis utilizing single-cell data implicates excitatory, inhibitory, and medium spiny neurons and the involvement of amygdala neurons in both mouse and human single-cell analyses. The associations are enriched for antidepressant targets and provide potential repurposing opportunities. Polygenic scores trained using European or multi-ancestry data predicted MD status across all ancestries, explaining up to 5.8% of MD liability variance in Europeans. These findings advance our global understanding of MD and reveal biological targets that may be used to target and develop pharmacotherapies addressing the unmet need for effective treatment.
Keywords: GWAS; depression; drugs; enrichment; genetic; genome-wide association study; neurons; pharmacotherapies; targets.
Copyright © 2024 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests C.M.L. is a member of the SAB for Myriad Neuroscience and has received consultancy fees from UCB.
Figures



References
-
- Howard DM, Adams MJ, Clarke T-K, Hafferty JD, Gibson J, Shirali M, Coleman JRI, Hagenaars SP, Ward J, Wigmore EM, et al. (2019). Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat. Neurosci. 22, 343–352. 10.1038/s41593-018-0326-7. - DOI - PMC - PubMed
-
- Major Depressive Disorder Working Group of the Psychiatric GWAS Consortium, Ripke S, Wray NR, Lewis CM, Hamilton SP, Weissman MM, Breen G, Byrne EM, Blackwood DHR, Boomsma DI, et al. (2013). A mega-analysis of genome-wide association studies for major depressive disorder. Mol. Psychiatry 18, 497–511. 10.1038/mp.2012.21. - DOI - PMC - PubMed
-
- Howard DM, Adams MJ, Shirali M, Clarke T-K, Marioni RE, Davies G, Coleman JRI, Alloza C, Shen X, Barbu MC, et al. (2018). Genome-wide association study of depression phenotypes in UK Biobank identifies variants in excitatory synaptic pathways. Nat. Commun. 9, 1470. 10.1038/s41467-018-03819-3. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
