

NADPH oxidases: redox regulation of cell homeostasis and disease
- PMID: 39814410
- PMCID: PMC12285607
- DOI: 10.1152/physrev.00034.2023
NADPH oxidases: redox regulation of cell homeostasis and disease
Abstract
The redox signaling network in mammals has garnered enormous interest and taken on major biological significance in recent years as the scope of NADPH oxidases (NOXs) as regulators of physiological signaling and cellular degeneration has grown exponentially. All NOX isoforms have in common the capacity to generate reactive oxygen species (ROS) superoxide anion (O2•-) and/or hydrogen peroxide (H2O2). A baseline, normal level of ROS formation supports a wide range of processes under physiological conditions. A disruption in redox balance caused by either the suppression or "super" induction of NOX off balance with antioxidant systems is associated with myriad diseases and cell/tissue damage. Over the past two to three decades, our understanding of NOXs has progressed from almost entirely a phagocyte-, antimicrobial-centered perspective to that of a family of enzymes that is vital to broad cellular function and organismal homeostasis. It is becoming increasingly evident that highly regulated, targeted oxidative protein modifications are elicited in a spatiotemporal manner and initiated at cell membranes in humans by seven NOX isoforms [NOXs 1, 2, 3, 4, 5 and dual oxidases (DUOXs) 1 and 2]. In a sense, this renders NOX-ROS signaling akin to that of other second messenger systems involving localized Ca2+ dynamics and tyrosine kinase transactivation. Accordingly, the study of ROS compartmentalization in subcellular organelles has been shown to be crucial to elucidating their role in cell phenotype modulation under physiological and pathophysiological conditions. The NOXs are as distinct in their distribution and activation as they are in their cellular functions, ranging from host defense, second messenger posttranslational modifications (PTMs) to transcriptional, epigenetic, and (de)differentiating effects. This review integrates past knowledge in the field with new focus areas on the leading edge of NOX-centered ROS signaling, including how a new wave of structural information provides insights for NOX biology and targeted therapies.
Keywords: NADPH oxidase; NOX; disease; physiology; redox signaling.
Conflict of interest statement
DISCLOSURES
The authors declare no conflicts of interest, financial or otherwise.
Figures


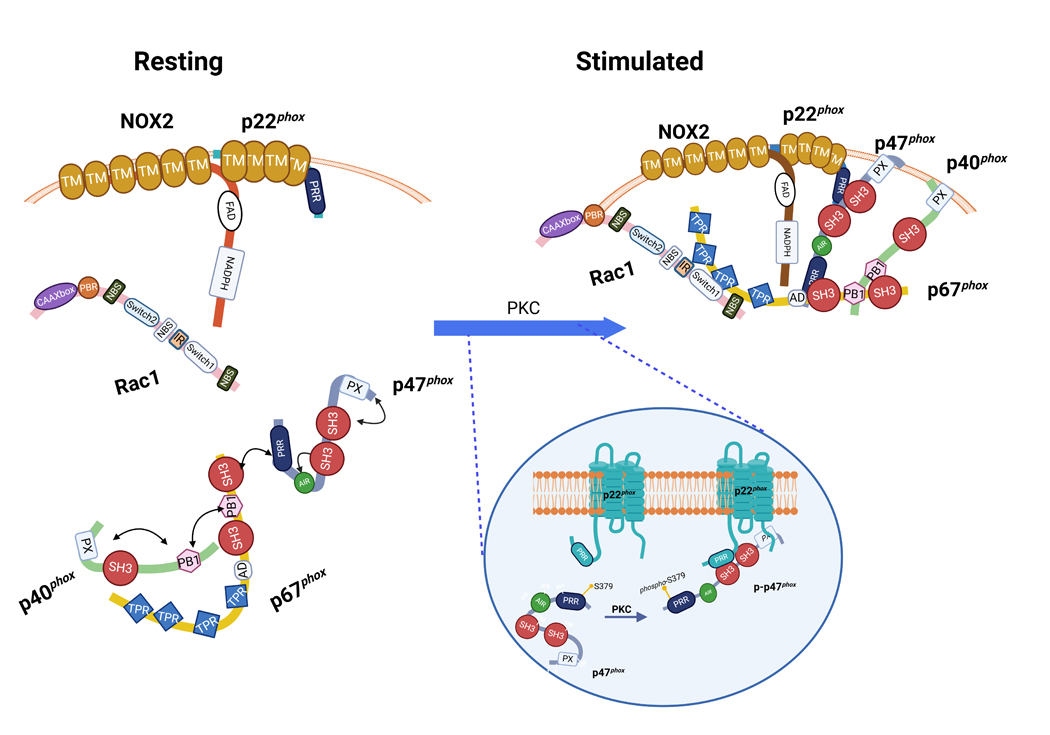











References
-
- Kehrer JP. The Haber-Weiss reaction and mechanisms of toxicity. Toxicology 149: 43–50, 2000. - PubMed
-
- Haber FWJ. Über die Katalyse des Hydroperoxydes Naturwissenschaften 20: 948–950, 1932.
Publication types
MeSH terms
Substances
Grants and funding
- R01 HL166985/HL/NHLBI NIH HHS/United States
- R21 AG089718/AG/NIA NIH HHS/United States
- 2020/12432-6/Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP)
- R01 HL142248/HL/NHLBI NIH HHS/United States
- T32 GM008424/GM/NIGMS NIH HHS/United States
- T32GM133332-02/HHS | National Institutes of Health (NIH)
- T32 GM133332/GM/NIGMS NIH HHS/United States
- R01HL142248-04/HHS | NIH | NHLBI | Division of Intramural Research (DIR)
- Vitalant & Western PA Blood Bank/University of Pittsburgh (Pitt)
- 1R01HL166985-01A1/HHS | NIH | National Heart, Lung, and Blood Institute (NHLBI)
LinkOut - more resources
Full Text Sources
Miscellaneous