

Massively parallel characterization of transcriptional regulatory elements
- PMID: 39814889
- PMCID: PMC11903340
- DOI: 10.1038/s41586-024-08430-9
Massively parallel characterization of transcriptional regulatory elements
Abstract
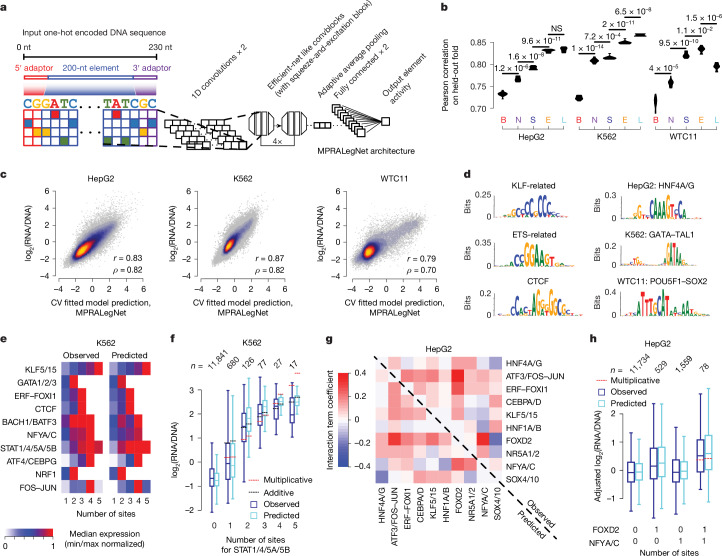
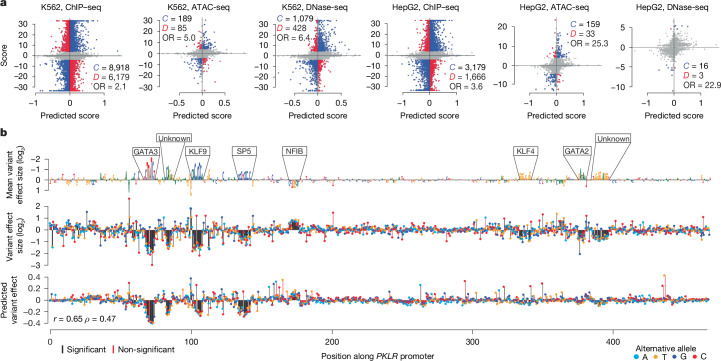
The human genome contains millions of candidate cis-regulatory elements (cCREs) with cell-type-specific activities that shape both health and many disease states1. However, we lack a functional understanding of the sequence features that control the activity and cell-type-specific features of these cCREs. Here we used lentivirus-based massively parallel reporter assays (lentiMPRAs) to test the regulatory activity of more than 680,000 sequences, representing an extensive set of annotated cCREs among three cell types (HepG2, K562 and WTC11), and found that 41.7% of these sequences were active. By testing sequences in both orientations, we find promoters to have strand-orientation biases and their 200-nucleotide cores to function as non-cell-type-specific 'on switches' that provide similar expression levels to their associated gene. By contrast, enhancers have weaker orientation biases, but increased tissue-specific characteristics. Utilizing our lentiMPRA data, we develop sequence-based models to predict cCRE function and variant effects with high accuracy, delineate regulatory motifs and model their combinatorial effects. Testing a lentiMPRA library encompassing 60,000 cCREs in all three cell types further identified factors that determine cell-type specificity. Collectively, our work provides an extensive catalogue of functional CREs in three widely used cell lines and showcases how large-scale functional measurements can be used to dissect regulatory grammar.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: V.A. is an employee of Sanofi, but performed this work independently of Sanofi. J.S. is a scientific advisory board member, consultant and/or co-founder of Cajal Neuroscience, Guardant Health, Maze Therapeutics, Camp4 Therapeutics, Phase Genomics, Adaptive Biotechnologies, Scale Biosciences, Somite Therapeutics, Sixth Street Capital and Pacific Biosciences. N.A. is a co-founder and on the scientific advisory board of Regel Therapeutics and receives funding from BioMarin Pharmaceutical Incorporated. All other authors declare no competing interests.
Figures













Update of
-
Massively parallel characterization of transcriptional regulatory elements in three diverse human cell types.bioRxiv [Preprint]. 2023 Mar 6:2023.03.05.531189. doi: 10.1101/2023.03.05.531189. bioRxiv. 2023. Update in: Nature. 2025 Mar;639(8054):411-420. doi: 10.1038/s41586-024-08430-9. PMID: 36945371 Free PMC article. Updated. Preprint.
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
