

Compound heterozygous mutations (p.L68R∗37 and p.T241N) lead to abnormal protein levels and structures in hereditary FVII deficiency
- PMID: 39817833
- PMCID: PMC11789605
- DOI: 10.1097/MBC.0000000000001340
Compound heterozygous mutations (p.L68R∗37 and p.T241N) lead to abnormal protein levels and structures in hereditary FVII deficiency
Abstract
Background: Congenital factor VII (FVII) deficiency is a genetic disorder characterized by decreased FVII activity, which sometimes leads to fatal bleeding. Numerous variants have been found in FVII deficiency, but mutations vary among patients. Each mutation deserves further exploration for each patient at risk of bleeding. We previously reported a Chinese patient with p.L68R∗37 and p.T241N compound heterozygous mutations. In this study, we further investigated the impact of these two mutations on the FVII expression through in vitro expression experiments.
Methods: Mutations were introduced into the FVII coding region using site-directed mutagenesis, and recombinant FVII was combined with two different plasmids, and then quantitative PCR and western blot analyses were performed subsequently.
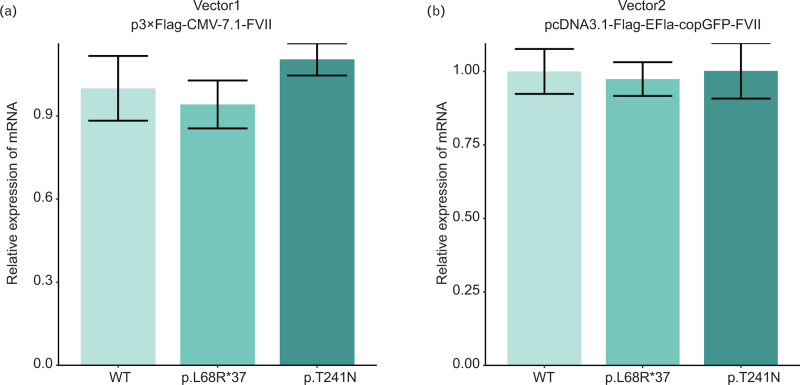
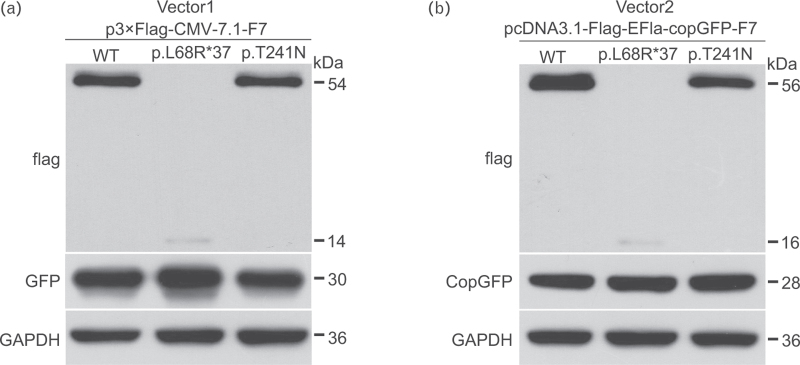
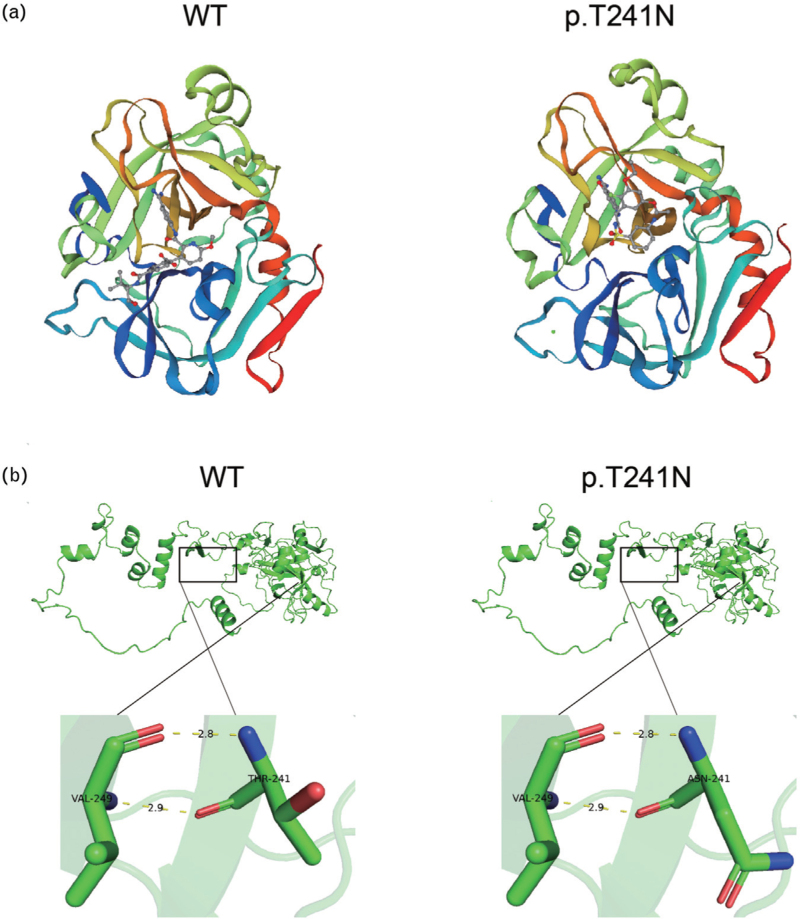
Results: The p.L68R∗37 mutation had no effect on mRNA levels but caused a significant decrease in protein levels. In the p.T241N mutant vector, mRNA levels did not show a noticeable decrease, but protein levels exhibited a slight decrease. Structural analysis revealed that the p.T241N mutation resulted in an altered secondary structure and protein instability, indicating impaired functional properties.
Conclusion: Our study demonstrated that the p.L68R∗37 and p.T241N mutations impacted the protein levels and function of FVII, ultimately leading to a severe reduction in FVII activity. This study may contribute to further understanding of the molecular pathogenesis of FVII deficiency and offer insights for genetic counseling.
Copyright © 2025 The Author(s). Published by Wolters Kluwer Health, Inc.
Conflict of interest statement
None of the authors has any conflict of interest to disclose.
Figures

Similar articles
-
Hereditary coagulation factor VII deficiency caused by novel compound heterozygous mutations c.572-1G>A and c.1037A>C in a Chinese pedigree.Gene. 2024 Nov 30;928:148788. doi: 10.1016/j.gene.2024.148788. Epub 2024 Jul 22. Gene. 2024. PMID: 39047958
-
A novel missense mutation close to the charge-stabilizing system in a patient with congenital factor VII deficiency.Blood Coagul Fibrinolysis. 2011 Jun;22(4):264-70. doi: 10.1097/MBC.0b013e3283447388. Blood Coagul Fibrinolysis. 2011. PMID: 21372693
-
[Novel double heterozygous mutations on Met306Val and Thr181Asn related to a hereditary coagulation factor VII deficiency].Zhonghua Yi Xue Za Zhi. 2006 Jan 10;86(2):124-7. Zhonghua Yi Xue Za Zhi. 2006. PMID: 16620721 Chinese.
-
A novel compound heterozygous mutation (c.64G > A and c.506-1G > A) associated with congenital coagulation factor VII deficiency: a case report and literature review.Ann Hematol. 2025 May;104(5):2995-3000. doi: 10.1007/s00277-025-06364-4. Epub 2025 Apr 21. Ann Hematol. 2025. PMID: 40257480 Free PMC article. Review.
-
Novel heterozygous F7 gene mutation (c. C1286T) associated with congenital factor VII deficiency: A case report and literature review.J Clin Lab Anal. 2022 May;36(5):e24349. doi: 10.1002/jcla.24349. Epub 2022 Mar 29. J Clin Lab Anal. 2022. PMID: 35349734 Free PMC article. Review.
References
-
- D’Andrea G, Bossone A, Lupone MR, Peyvandi F, Maisto G, Perricone F, et al. . Molecular characterization of a factor VII deficient patient supports the importance of the second epidermal growth factor-like domain. Haematologica 2004; 89:979–984. - PubMed
-
- Mannucci PM, Tuddenham EG. The hemophilias--from royal genes to gene therapy. N Engl J Med 2001; 344:1773–1779. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
