

Signalling pathways involved in urotensin II induced ventricular myocyte hypertrophy
- PMID: 39820183
- PMCID: PMC11737703
- DOI: 10.1371/journal.pone.0313119
Signalling pathways involved in urotensin II induced ventricular myocyte hypertrophy
Abstract
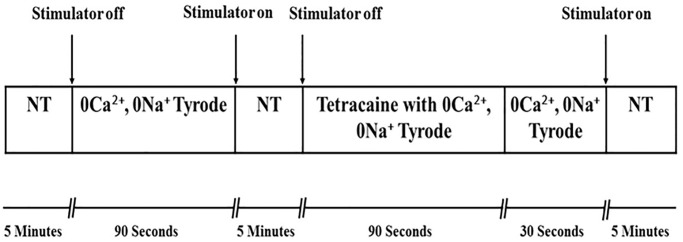
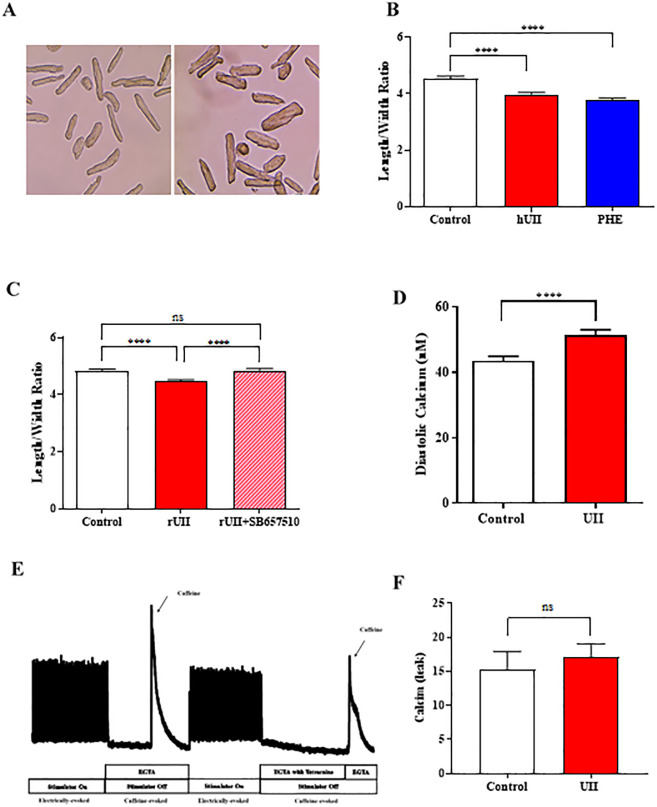
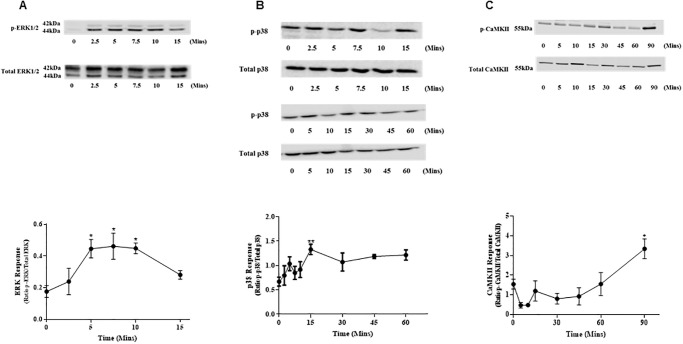
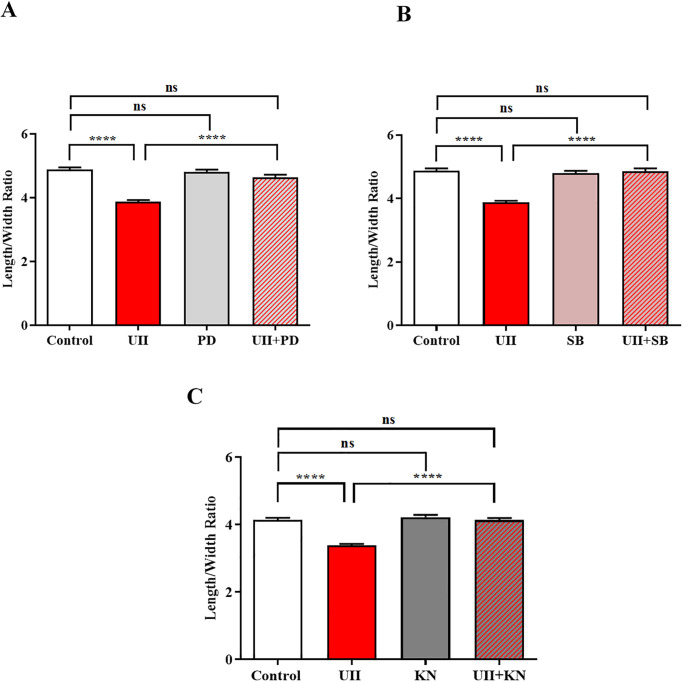
Sustained pathologic myocardial hypertrophy can result in heart failure(HF); a significant health issue affecting a large section of the population worldwide. In HF there is a marked elevation in circulating levels of the peptide urotensin II(UII) but it is unclear whether this is a result of hypertrophy or whether the high levels contribute to the development of hypertrophy. The aim of this study is to investigate a role of UII and its receptor UT in the development of cardiac hypertrophy and the signalling molecules involved. Ventricular myocytes isolated from adult rat hearts were treated with 200nM UII for 48hours and hypertrophy was quantified from measurements of length/width (L/W) ratio. UII resulted in a change in L/W ratio from 4.53±0.10 to 3.99±0.06; (p<0.0001) after 48hours. The response is reversed by the UT-antagonist SB657510 (1μM). UT receptor activation by UII resulted in the activation of ERK1/2, p38 and CaMKII signalling pathways measured by Western blotting; these are involved in the induction of hypertrophy. JNK was not involved. Moreover, ERK1/2, P38 and CaMKII inhibitors completely blocked UII-induced hypertrophy. Sarcoplasmic reticulum (SR) Ca2+-leak was investigated in isolated myocytes. There was no significant increase in SR Ca2+-leak. Our results suggest that activation of MAPK and CaMKII signalling pathways are involved in the hypertrophic response to UII. Collectively our data suggest that increased circulating UII may contribute to the development of left ventricular hypertrophy and pharmacological inhibition of the UII/UT receptor system may prove beneficial in reducing adverse remodeling and alleviating contractile dysfunction in heart disease.
Copyright: © 2025 Al Ali et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
