

Contribution of hypoxia-inducible factor 1alpha to pathogenesis of sarcomeric hypertrophic cardiomyopathy
- PMID: 39820339
- PMCID: PMC11739497
- DOI: 10.1038/s41598-025-85187-9
Contribution of hypoxia-inducible factor 1alpha to pathogenesis of sarcomeric hypertrophic cardiomyopathy
Abstract
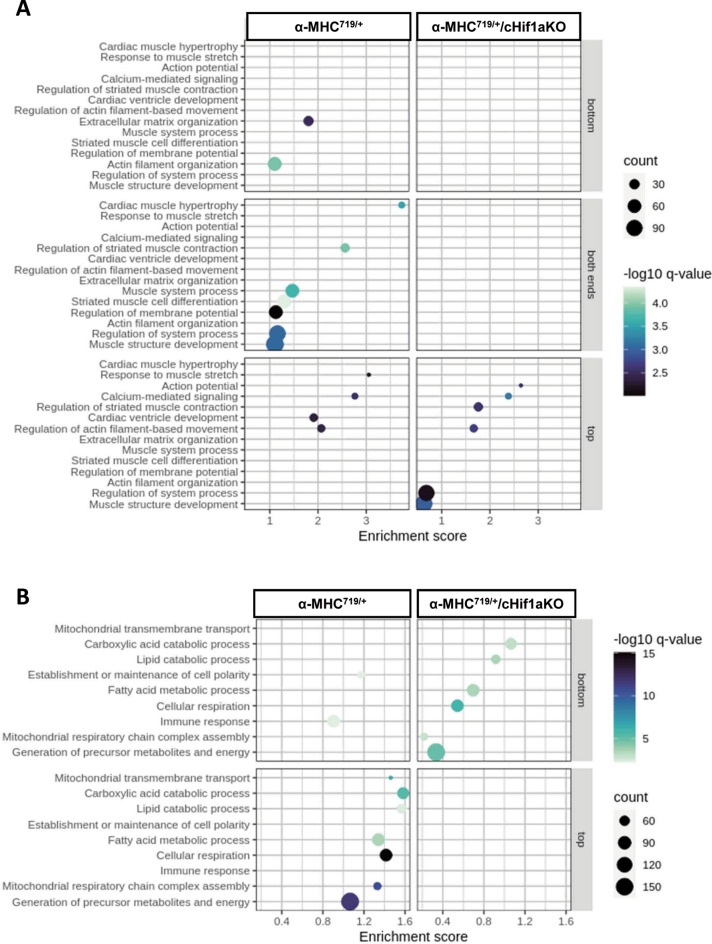
Hypertrophic cardiomyopathy (HCM) caused by autosomal-dominant mutations in genes coding for structural sarcomeric proteins, is the most common inherited heart disease. HCM is associated with myocardial hypertrophy, fibrosis and ventricular dysfunction. Hypoxia-inducible transcription factor-1α (Hif-1α) is the central master regulators of cellular hypoxia response and associated with HCM. Yet its exact role remains to be elucidated. Therefore, the effect of a cardiomyocyte-specific Hif-1a knockout (cHif1aKO) was studied in an established α-MHC719/+ HCM mouse model that exhibits the classical features of human HCM. The results show that Hif-1α protein and HIF targets were upregulated in left ventricular tissue of α-MHC719/+ mice. Cardiomyocyte-specific abolishment of Hif-1a blunted the disease phenotype, as evidenced by decreased left ventricular wall thickness, reduced myocardial fibrosis, disordered SRX/DRX state and ROS production. cHif1aKO induced normalization of pro-hypertrophic and pro-fibrotic left ventricular remodeling signaling evidenced on whole transcriptome and proteomics analysis in α-MHC719/+ mice. Proteomics of serum samples from patients with early onset HCM revealed significant modulation of HIF. These results demonstrate that HIF signaling is involved in mouse and human HCM pathogenesis. Cardiomyocyte-specific knockout of Hif-1a attenuates disease phenotype in the mouse model. Targeting Hif-1α might serve as a therapeutic option to mitigate HCM disease progression.
Keywords: HIF1A; Hypertrophic cardiomyopathy; Hypertrophy; Hypoxia; Myocardial fibrosis.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Competing interests: Wolf CM: Honoraria: Novo Nordisk and Bristol-Myers Squibb; Dr. Wolf is a consultant for Day One Biopharmaceuticals, Inc., BioMarin Pharmaceuticals, Adrenomed AG, and Pliant Therapeutics. Dr. Wolf has ownership interest (Preventage Therapeutics). There are no other competing financial and non-financial interests
Figures







References
-
- Maron, B. J. et al. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA study. Coronary artery risk development in (young) adults. Circulation92, 785–789. 10.1161/01.cir.92.4.785 (1995). - PubMed
-
- Maron, B. J. Contemporary insights and strategies for risk stratification and prevention of sudden death in hypertrophic cardiomyopathy. Circulation121, 445–456. 10.1161/CIRCULATIONAHA.109.878579 (2010). - PubMed
-
- Watkins, H. Sudden death in hypertrophic cardiomyopathy. N. Engl. J. Med.342, 422–424. 10.1056/NEJM200002103420609 (2000). - PubMed
-
- Geisterfer-Lowrance, A. A. et al. A molecular basis for familial hypertrophic cardiomyopathy: a beta cardiac myosin heavy chain gene missense mutation. Cell62, 999–1006. 10.1016/0092-8674(90)90274-i (1990). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
