

Challenging the Astral mass analyzer to quantify up to 5,300 proteins per single cell at unseen accuracy to uncover cellular heterogeneity
- PMID: 39820751
- PMCID: PMC11903296
- DOI: 10.1038/s41592-024-02559-1
Challenging the Astral mass analyzer to quantify up to 5,300 proteins per single cell at unseen accuracy to uncover cellular heterogeneity
Abstract
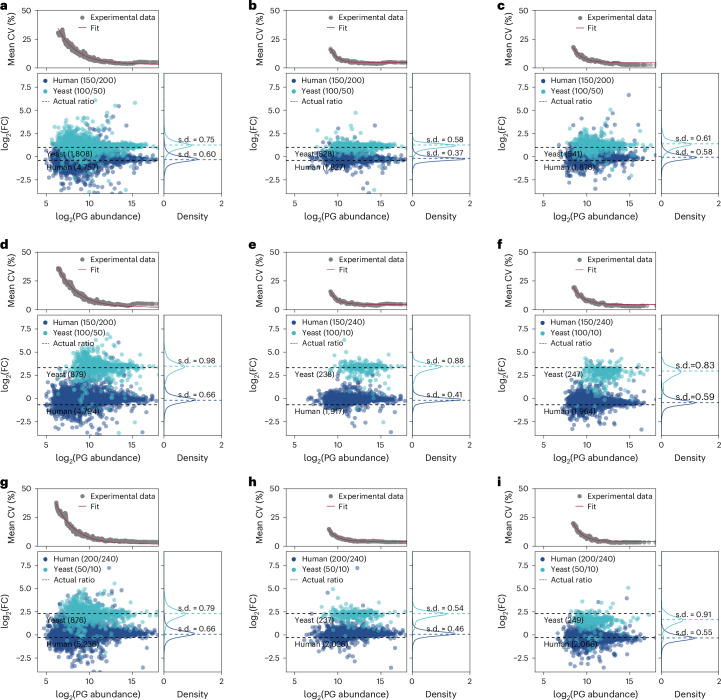
Despite significant advancements in sample preparation, instrumentation and data analysis, single-cell proteomics is currently limited by proteomic depth and quantitative performance. Here we demonstrate highly improved depth of proteome coverage as well as accuracy and precision for quantification of ultra-low input amounts. Using a tailored library, we identify up to 7,400 protein groups from as little as 250 pg of HeLa cell peptides at a throughput of 50 samples per day. Using a two-proteome mix, we check for optimal parameters of quantification and show that fold change differences of 2 can still be successfully determined at single-cell-level inputs. Eventually, we apply our workflow to A549 cells, yielding a proteome coverage ranging from 1,801 to a maximum of >5,300 protein groups from a single cell depending on cell size and search strategy used, which allows for the study of dependencies between cell size and cell cycle phase. Additionally, our workflow enables us to distinguish between in vitro analogs of two human blastocyst lineages: naive human pluripotent stem cells (epiblast) and trophectoderm-like cells. Our data harmoniously align with transcriptomic data, indicating that single-cell proteomics possesses the capability to identify biologically relevant differences within the blastocyst.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: T.N.A., E.D. and B.D. are employees of Thermo Fisher Scientific. The other authors declare no competing interests.
Figures






References
MeSH terms
Substances
Grants and funding
- LS20-079/Vienna Science and Technology Fund (Wiener Wissenschafts-, Forschungs- und Technologiefonds)
- P35045-B/Austrian Science Fund (Fonds zur Förderung der Wissenschaftlichen Forschung)
- F 8801-B/Austrian Science Fund (Fonds zur Förderung der Wissenschaftlichen Forschung)
- ESP497/Austrian Science Fund (Fonds zur Förderung der Wissenschaftlichen Forschung)
- 2022/01 AT-SCP/Österreichische Forschungsförderungsgesellschaft (Austrian Research Promotion Agency)
LinkOut - more resources
Full Text Sources
Medical
