

Design and validation of cell-based potency assays for frataxin supplementation treatments
- PMID: 39823061
- PMCID: PMC11735916
- DOI: 10.1016/j.omtm.2024.101347
Design and validation of cell-based potency assays for frataxin supplementation treatments
Abstract
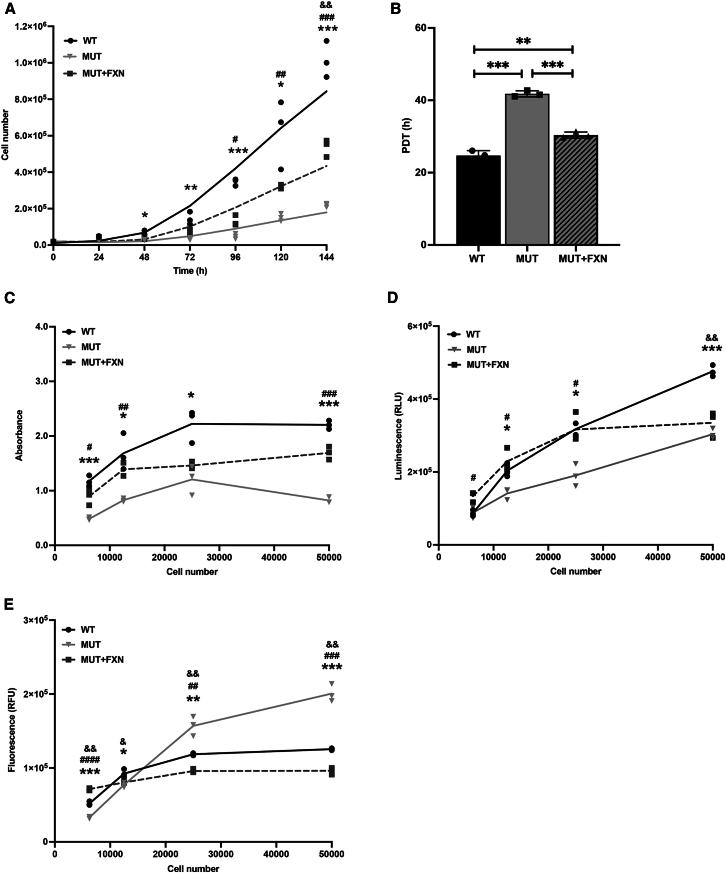
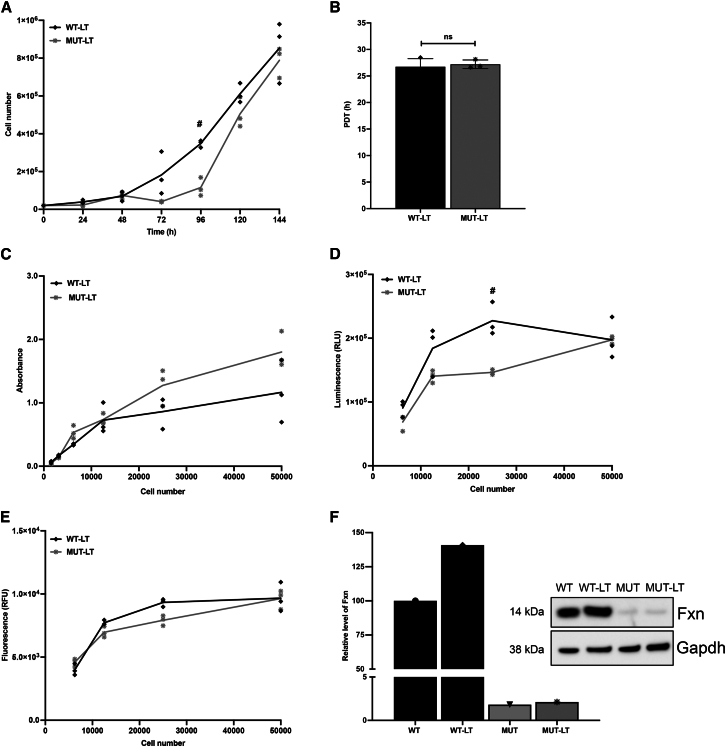
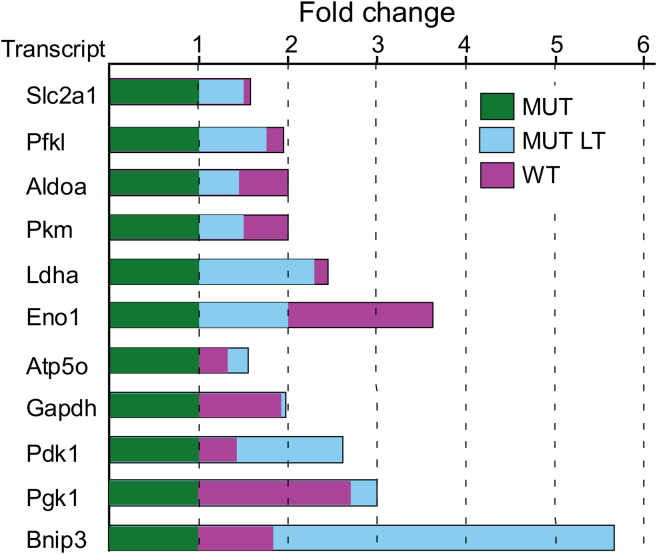
Friedreich's ataxia (FRDA) is a multisystem, autosomal recessive disorder caused by mutations in the frataxin (FXN) gene. As FRDA is considered an FXN deficiency disorder, numerous therapeutic approaches in development or clinical trials aim to supplement FXN or restore endogenous FXN expression. These include gene therapy, protein supplementation, genome editing or upregulation of FXN transcription. To evaluate efficacy of these therapies, potency assays capable of quantitative determination of FXN biological activity are needed. Herein, we evaluate the suitability of mouse embryonic fibroblasts derived from Fxn G127V knockin mice (MUT MEFs) as a candidate for cell-based potency assays. We demonstrate that these cells, when immortalized, continue to express minute amounts of Fxn and exhibit a broad range of phenotypes that result from severe Fxn deficiency. Exogenous FXN supplementation reverses these phenotypes. Thus, immortalized MUT MEFs are an excellent tool for developing potency assays to validate novel FRDA therapies. Care needs to be exercised while utilizing these cell lines, as extended passaging results in molecular changes that spontaneously reverse FRDA-like phenotypes without increasing Fxn expression. Based on transcriptome analyses, we identified the Warburg effect as the mechanism allowing cells expressing a minimal level of Fxn to thrive under standard cell culture conditions.
Keywords: Friedreich’s ataxia; aerobic glycolysis; frataxin; gene therapy; potency assay.
© 2024 The Author(s).
Conflict of interest statement
E.S. is an employee of Friedreich’s Ataxia Research Alliance, and M.N. and R.B.W. are members of the Friedreich’s Ataxia Research Alliance scientific advisory board.
Figures





Similar articles
-
Mitochondrial damage and senescence phenotype of cells derived from a novel frataxin G127V point mutation mouse model of Friedreich's ataxia.Dis Model Mech. 2020 Jul 27;13(7):dmm045229. doi: 10.1242/dmm.045229. Dis Model Mech. 2020. PMID: 32586831 Free PMC article.
-
A Comprehensive Transcriptome Analysis Identifies FXN and BDNF as Novel Targets of miRNAs in Friedreich's Ataxia Patients.Mol Neurobiol. 2020 Jun;57(6):2639-2653. doi: 10.1007/s12035-020-01899-1. Epub 2020 Apr 14. Mol Neurobiol. 2020. PMID: 32291635 Free PMC article.
-
Neurobehavioral deficits of mice expressing a low level of G127V mutant frataxin.Neurobiol Dis. 2023 Feb;177:105996. doi: 10.1016/j.nbd.2023.105996. Epub 2023 Jan 10. Neurobiol Dis. 2023. PMID: 36638893 Free PMC article.
-
Recent Advances in the Elucidation of Frataxin Biochemical Function Open Novel Perspectives for the Treatment of Friedreich's Ataxia.Front Neurosci. 2022 Mar 2;16:838335. doi: 10.3389/fnins.2022.838335. eCollection 2022. Front Neurosci. 2022. PMID: 35310092 Free PMC article. Review.
-
Advantages and Limitations of Gene Therapy and Gene Editing for Friedreich's Ataxia.Front Genome Ed. 2022 May 17;4:903139. doi: 10.3389/fgeed.2022.903139. eCollection 2022. Front Genome Ed. 2022. PMID: 35663795 Free PMC article. Review.
References
-
- Campuzano V., Montermini L., Moltò M.D., Pianese L., Cossée M., Cavalcanti F., Monros E., Rodius F., Duclos F., Monticelli A., et al. Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271:1423–1427. - PubMed
-
- Cossee M., Durr A., Schmitt M., Dahl N., Trouillas P., Allinson P., Kostrzewa M., Nivelon-Chevallier A., Gustavson K.H., Kohlschutter A., et al. Friedreich's ataxia: point mutations and clinical presentation of compound heterozygotes. Ann. Neurol. 1999;45:200–206. - PubMed
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
