

Somatic CAG repeat expansion in blood associates with biomarkers of neurodegeneration in Huntington's disease decades before clinical motor diagnosis
- PMID: 39825149
- PMCID: PMC11922752
- DOI: 10.1038/s41591-024-03424-6
Somatic CAG repeat expansion in blood associates with biomarkers of neurodegeneration in Huntington's disease decades before clinical motor diagnosis
Abstract
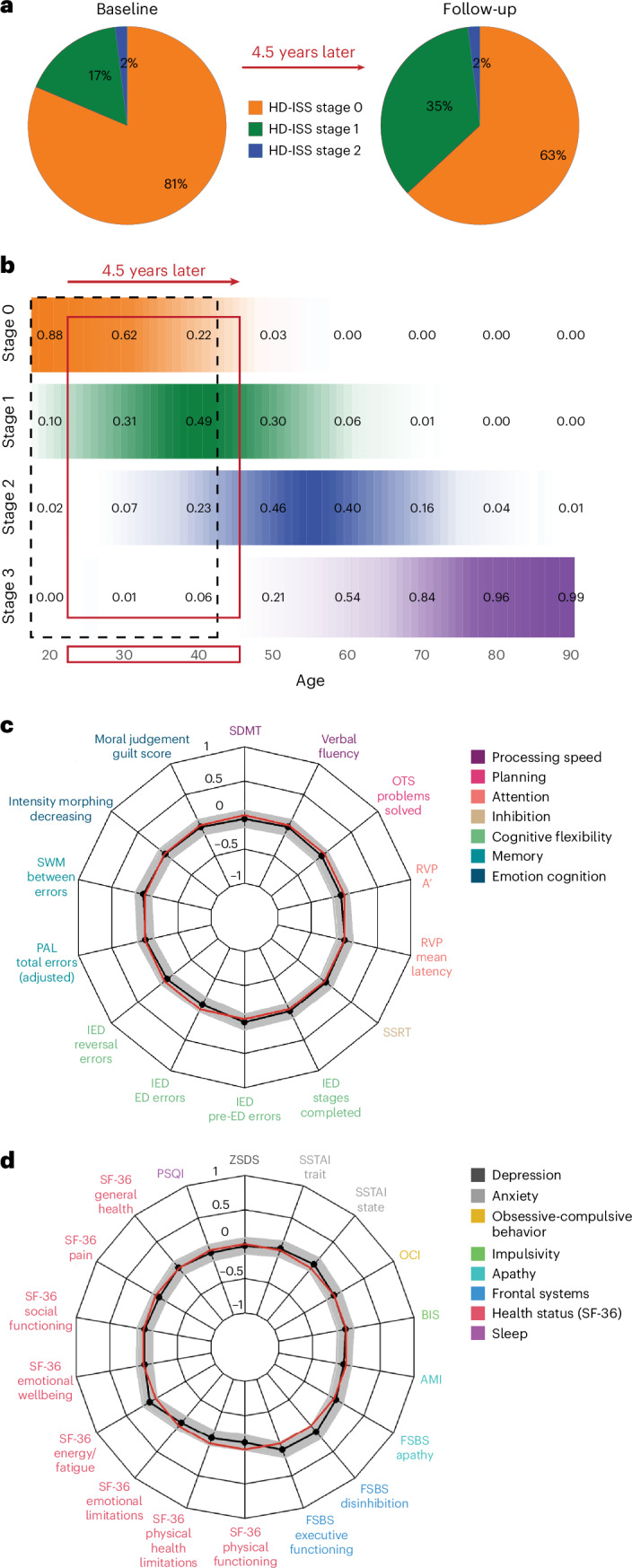
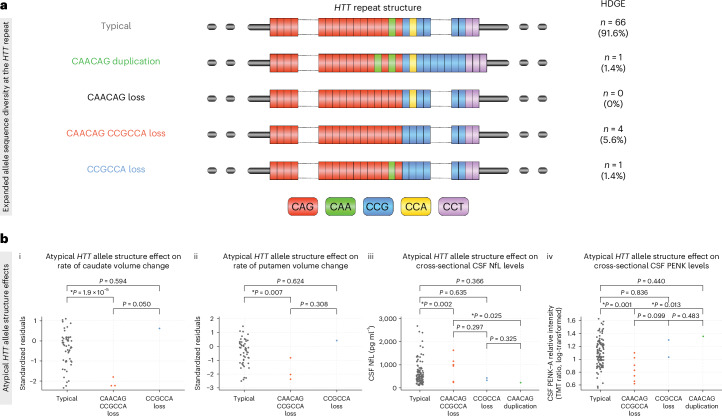
Huntington's disease (HD) is an autosomal dominant neurodegenerative disease with the age at which characteristic symptoms manifest strongly influenced by inherited HTT CAG length. Somatic CAG expansion occurs throughout life and understanding the impact of somatic expansion on neurodegeneration is key to developing therapeutic targets. In 57 HD gene expanded (HDGE) individuals, ~23 years before their predicted clinical motor diagnosis, no significant decline in clinical, cognitive or neuropsychiatric function was observed over 4.5 years compared with 46 controls (false discovery rate (FDR) > 0.3). However, cerebrospinal fluid (CSF) markers showed very early signs of neurodegeneration in HDGE with elevated neurofilament light (NfL) protein, an indicator of neuroaxonal damage (FDR = 3.2 × 10-12), and reduced proenkephalin (PENK), a surrogate marker for the state of striatal medium spiny neurons (FDR = 2.6 × 10-3), accompanied by brain atrophy, predominantly in the caudate (FDR = 5.5 × 10-10) and putamen (FDR = 1.2 × 10-9). Longitudinal increase in somatic CAG repeat expansion ratio (SER) in blood was a significant predictor of subsequent caudate (FDR = 0.072) and putamen (FDR = 0.148) atrophy. Atypical loss of interruption HTT repeat structures, known to predict earlier age at clinical motor diagnosis, was associated with substantially faster caudate and putamen atrophy. We provide evidence in living humans that the influence of CAG length on HD neuropathology is mediated by somatic CAG repeat expansion. These critical mechanistic insights into the earliest neurodegenerative changes will inform the design of preventative clinical trials aimed at modulating somatic expansion. ClinicalTrials.gov registration: NCT06391619 .
© 2025. The Author(s).
Conflict of interest statement
Competing interests: J.B.R. is supported by the Wellcome Trust (220258), the Medical Research Council (MC_UU_00030/14; MR/T033371/1) and the NIHR Cambridge Biomedical Research Centre (NIHR203312). The views expressed are those of the authors and not necessarily those of the NIHR or the Department of Health and Social Care. J.B.R. has undertaken paid consultancy for Asceneuron, Astronautx, Astex, ClinicalInk, CumulusNeuro, Curasen, Invivro, Prevail and SVHealth; received research funding unrelated to the current work from AstraZeneca, Lily, GSK and Janssen; and is Chief Scientific Advisor to Alzheimer’s Research UK. D.G.M. has been a scientific consultant and/or received honoraria/grants from AMO Pharma, Dyne, F. Hoffman-La Roche, Function Rx, LoQus23, MOMA Therapeutics, Novartis, Ono Pharmaceuticals, Pfizer Pharmaceuticals, Rgenta Therapeutics, Sanofi and Sarepta Therapeutics. J.G. is supported by Alzheimerfonden (AF-980746) and Stiftelsen för Gamla tjänarinnor (2022-01324). J.B.R. has appeared as an expert witness to the Medicines and Healthcare products Regulatory Agency, unrelated to the current work. D.G.M. is on the Scientific Advisory Board of the Myotonic Dystrophy Foundation and EuroDyMA (European Dystrophia Myotonica Association), is a scientific advisor to the Myotonic Dystrophy Support Group and is a vice president for research of Muscular Dystrophy UK. G.R. is a nonexecutive director of UCL Business. D.R.L. is an unpaid academic member of the Critical Path Institute HD-RSC Consortium Coordinating Committee. B.J.S. is a co-inventor of the Cambridge Neuropsychological Test Automated Battery. I.B.M. is an associate editor for Frontiers in Neurology. The other authors declare no competing interests.
Figures








Comment in
-
Degeneration decades before disease onset.Nat Rev Neurol. 2025 Mar;21(3):125. doi: 10.1038/s41582-025-01069-8. Nat Rev Neurol. 2025. PMID: 39962296 No abstract available.
References
-
- Vonsattel, J. P. et al. Neuropathological classification of Huntington’s disease. J. Neuropathol. Exp. Neurol.44, 559–577 (1985). - PubMed
-
- Kennedy, L. et al. Dramatic tissue-specific mutation length increases are an early molecular event in Huntington disease pathogenesis. Hum. Mol. Genet.12, 3359–3367 (2003). - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
