

Genomic reanalysis of a pan-European rare-disease resource yields new diagnoses
- PMID: 39825153
- PMCID: PMC11835725
- DOI: 10.1038/s41591-024-03420-w
Genomic reanalysis of a pan-European rare-disease resource yields new diagnoses
Erratum in
-
Publisher Correction: Genomic reanalysis of a pan-European rare-disease resource yields new diagnoses.Nat Med. 2025 Aug;31(8):2819-2820. doi: 10.1038/s41591-025-03754-z. Nat Med. 2025. PMID: 40537530 Free PMC article. No abstract available.
Abstract
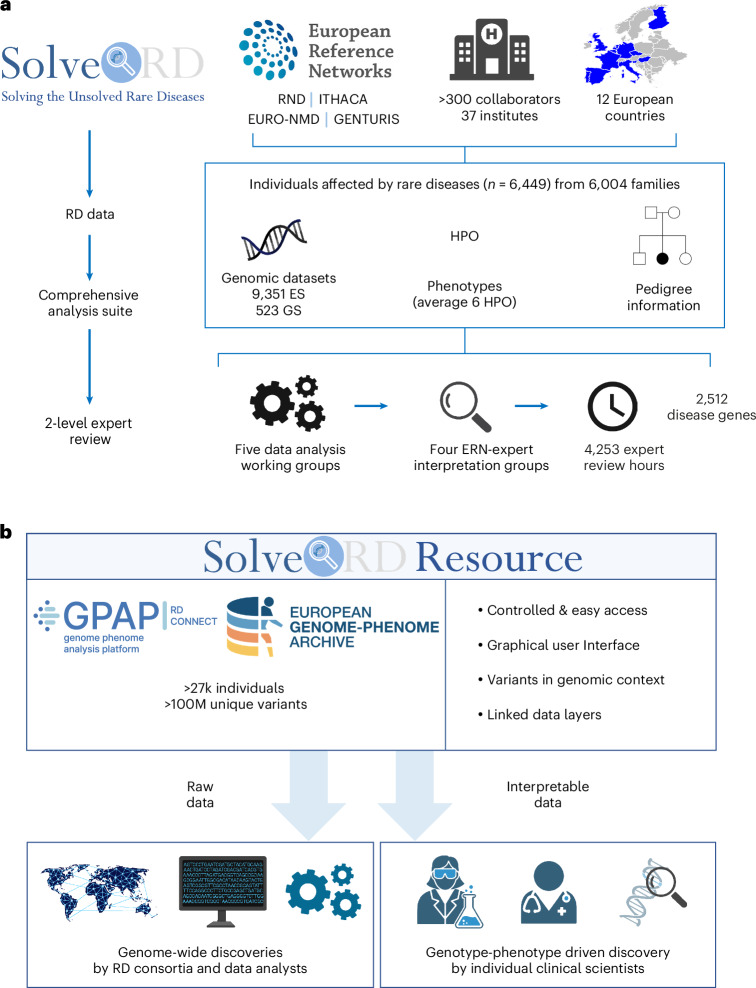
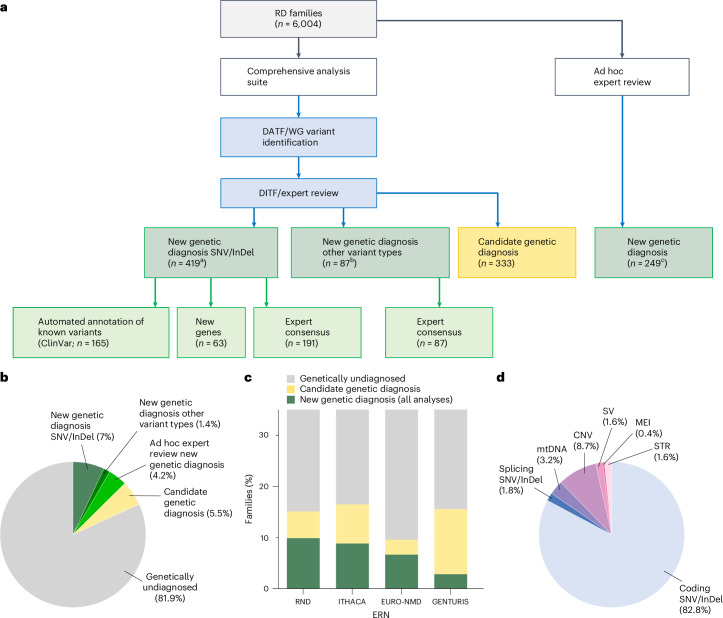
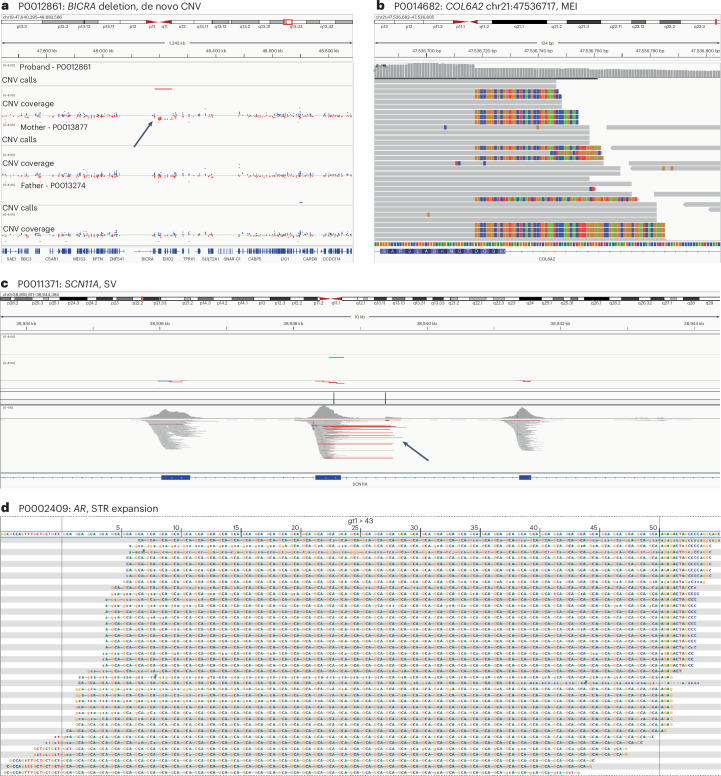
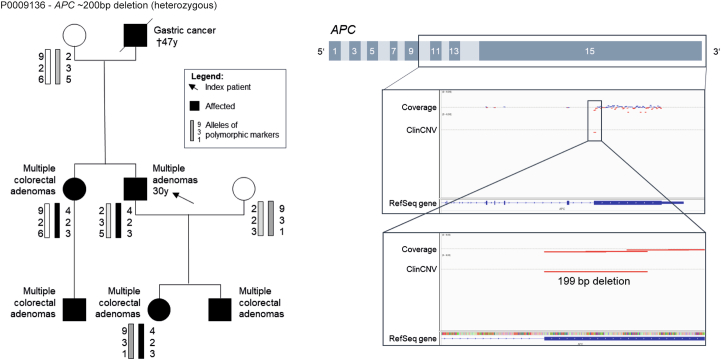
Genetic diagnosis of rare diseases requires accurate identification and interpretation of genomic variants. Clinical and molecular scientists from 37 expert centers across Europe created the Solve-Rare Diseases Consortium (Solve-RD) resource, encompassing clinical, pedigree and genomic rare-disease data (94.5% exomes, 5.5% genomes), and performed systematic reanalysis for 6,447 individuals (3,592 male, 2,855 female) with previously undiagnosed rare diseases from 6,004 families. We established a collaborative, two-level expert review infrastructure that allowed a genetic diagnosis in 506 (8.4%) families. Of 552 disease-causing variants identified, 464 (84.1%) were single-nucleotide variants or short insertions/deletions. These variants were either located in recently published novel disease genes (n = 67), recently reclassified in ClinVar (n = 187) or reclassified by consensus expert decision within Solve-RD (n = 210). Bespoke bioinformatics analyses identified the remaining 15.9% of causative variants (n = 88). Ad hoc expert review, parallel to the systematic reanalysis, diagnosed 249 (4.1%) additional families for an overall diagnostic yield of 12.6%. The infrastructure and collaborative networks set up by Solve-RD can serve as a blueprint for future further scalable international efforts. The resource is open to the global rare-disease community, allowing phenotype, variant and gene queries, as well as genome-wide discoveries.
© 2025. The Author(s).
Conflict of interest statement
Competing interests: M. Synofzik has received consultancy honoraria from Janssen, Ionis, Orphazyme, Servier, Reata, GenOrph and AviadoBio, all unrelated to the present manuscript. B.v.d.W. has received consultancy honoraria from, and/or has served on advisory boards for, Servier, Biohaven Pharmaceuticals, Vico Therapeutics and Biogen, all unrelated to the present manuscript. The other authors declare no competing interests.
Figures









References
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
