

Intercellular TIMP-1-CD63 signaling directs the evolution of immune escape and metastasis in KRAS-mutated pancreatic cancer cells
- PMID: 39825392
- PMCID: PMC11742192
- DOI: 10.1186/s12943-024-02207-4
Intercellular TIMP-1-CD63 signaling directs the evolution of immune escape and metastasis in KRAS-mutated pancreatic cancer cells
Abstract
Background and aims: Oncogenic KRAS mutations are present in approximately 90% of pancreatic ductal adenocarcinoma (PDAC). However, Kras mutation alone is insufficient to transform precancerous cells into metastatic PDAC. This study investigates how KRAS-mutated epithelial cells acquire the capacity to escape senescence or even immune clearance, thereby progressing to advanced PDAC.
Methods: Single-cell RNA sequencing and analysis of primary PDAC tumors were conducted. Genetically engineered pancreas-specific Kras-mutated, dual specificity phosphatase-2 (Dusp2) knockout mouse models were established. Human and mouse primary pancreatic cancer cell lines were used for in vitro assessment of cancer characteristics. Tumor progression was studied via pancreas orthotopic and portal vein injection in the immune-competent mice. Clinical relevance was validated by digital spatial transcriptomic analysis of PDAC tumors.
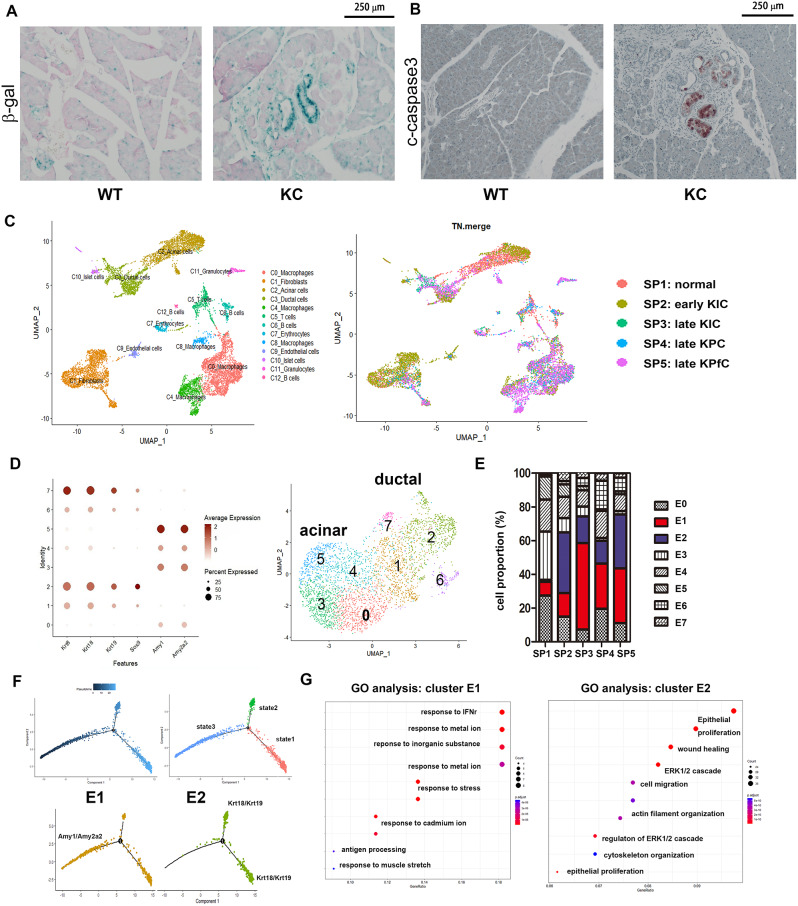
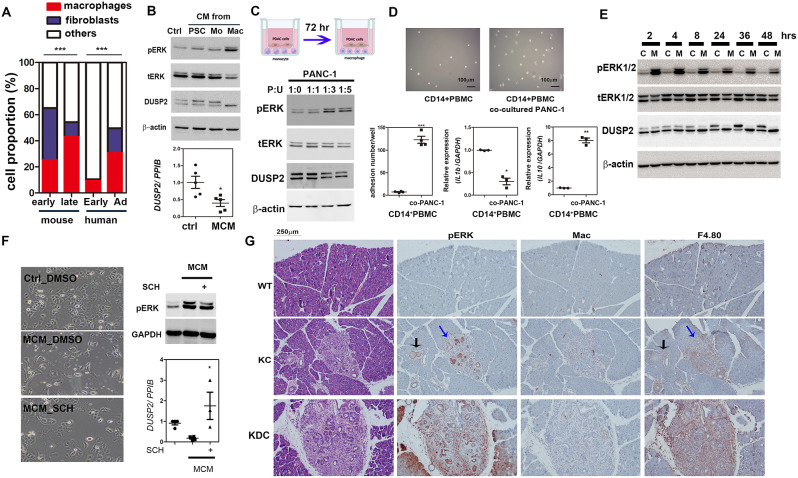
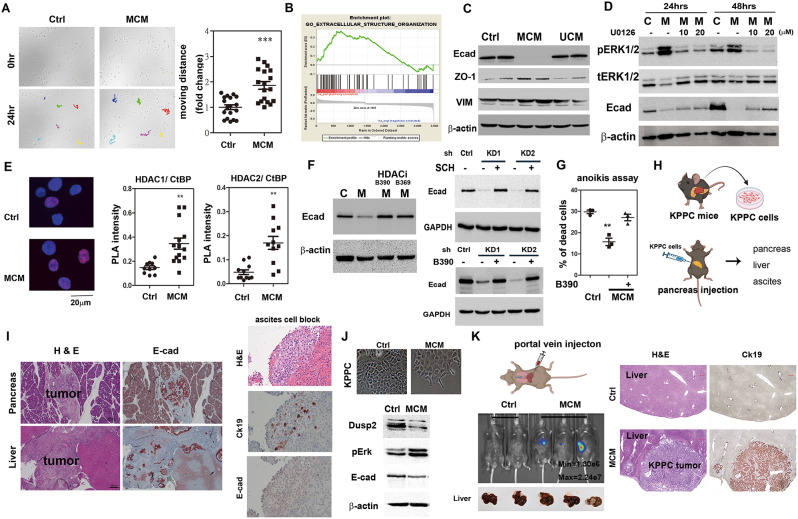
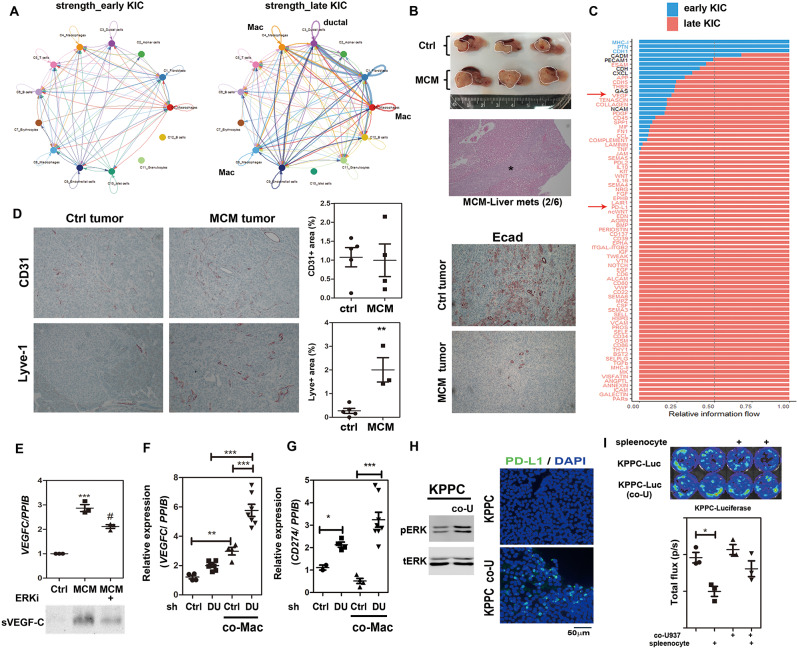
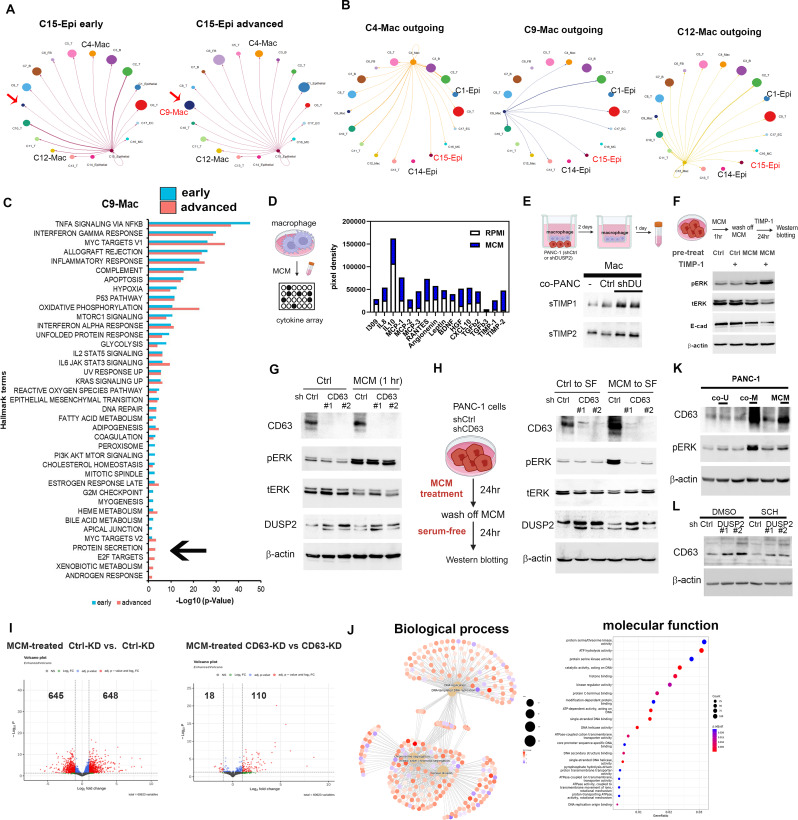
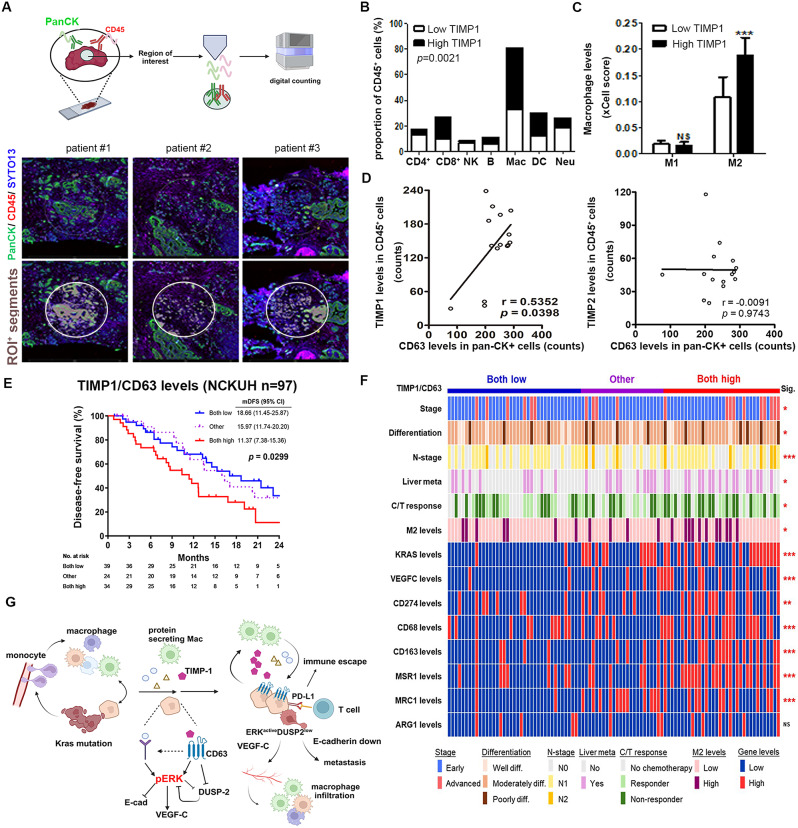
Results: Kras mutation induces the formation of pancreatic intraepithelial neoplasia (PanIN), these lesions also exhibit significant apoptotic signals. Single-cell RNA sequencing identified a subset of ERKactiveDUSP2low cells continuing to expand from early to advanced stage PDAC. In vitro and in vivo studies reveal that early infiltrating macrophage-derived tissue inhibitor of metallopeptidase 1 (TIMP-1) is the key factor in maintaining the ERKactiveDUSP2low cell population in a CD63-dependent manner. The ERKactiveDUSP2low cancer cells further exacerbate macrophage-mediated cancer malignancy, including loss of epithelial trait, increased lymphangiogenesis, and immune escape. Digital spatial profiling analysis of PDAC samples demonstrates the colocalization of TIMP-1high macrophages and CD63high cancer cells. The presence of TIMP-1high macrophages and CD63high epithelial cells correlates with poor prognosis in PDAC.
Conclusions: Our study reveals the vicious cycle between early infiltrating macrophages and pancreatic cancer cells, providing a mechanistic insight into the dynamic regulation directing pancreatic cancer progression.
© 2025. The Author(s).
Conflict of interest statement
Declarations. Ethical approval: Experimental procedures of animal studies were approved by the Institutional Animal Care and Use committee at the National Chung Kung University. Tumor specimens were obtained from PDAC patients undergoing surgical resection at the National Cheng Kung University Hospital (NCKUH) under Institutional Review Board (IRB)-approved protocol (IRB number: NCKUH B-ER-110-420). Competing interests: The authors declare no competing interests.
Figures

References
-
- Hingorani SR, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7(5):469–83. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
